
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Photo-patterned multi-domain multi-component hybrid hydrogels†
Daniel J.
Cornwell
and
David K.
Smith
 *
*
Department of Chemistry, University of York, Heslington, York, YO10 5DD, UK. E-mail: david.smith@york.ac.uk
First published on 26th May 2020
Abstract
This paper explores multi-component gelation systems containing two low-molecular-weight gelators and a polymer gelator. By controlled spatial and temporal application of different triggers – physical and chemical – it is possible to sequentially assemble gel networks, with a degree of self-sorting. A photo-patterned gel with four different domains was fabricated from a complex mixture of components, with the history of each domain programming the gel.
Hydrogels are soft materials with potential applications ranging from nanoscale electronics to regenerative medicine.1 Such gels fall into two fundamental classes, those based on polymer gelators (PGs),2 in which crosslinked or entangled polymer chains constitute a ‘solid-like’ network, and those in which low-molecular-weight gelators (LMWGs) self-assemble into an extended network.3 In general, PGs form more robust gels, while LMWGs, although more programmable and responsive, generally form weaker gels. An emerging strategy to tailor the performance of such gels employs multiple gelators in a single integrated material – a ‘multi-component gel’.4 Two different LMWGs can self-sort, co-assemble or disrupt one another's assembly. Self-sorting can yield gels in which orthogonal networks provide different properties.5 For example, it is possible to combine a responsive LMWG with a robust PG.6
For high-tech applications, the ability to pattern and shape gels can add significant value,7 but the relatively poor rheological performance of LMWGs can make this challenging. There is scope to pattern gels with different domains, each programmed with different chemical information. Photo-patterning has emerged as a powerful approach, with light driving the assembly of photo-responsive gelators,8 or disassembling such gels, etching away part of the overall structure.9 Combining two gelators can give greater control over photo-patterning, as a pre-existing gel network can limit convection and diffusion and hence improve spatial resolution. In 2015, Adams and co-workers demonstrated that a photo-responsive LMWG network could be disassembled in the presence of another (‘etching away’),10 and we reported the assembly of a photo-responsive LMWG in the presence of another (‘writing in’).11 A number of other reports have used two-components to create ‘multi-domain’ gels.12
With the target of programming structured soft matter systems for high-tech applications, we became interested in the challenge of assembling patterned gels from complex systems with more components. Here, we report preliminary results combining three gelators (two LMWGs and one PG). Previously, we reported that DBS-CO2H (pKa = 5.4) and DBS-Gly (pKa = 4.3) partly self-sort on acid-induced assembly as a result of their different pKa values (Fig. 1).11 DBS-CO2H and PEGDM (Fig. 1) can also be orthogonally assembled via pH change and photo-activation respectively.13 Here, we aimed to combine all three gelators in an integrated gel. The goal was to achieve sequential assembly of the gelators using three triggers: (i) 2-hydroxy-4′-(2-hydroxyethoxy)-2-methylpropiophenone (HMMP) is the photo-initiator (PI) for PEGDM polymerisation, (ii) glucono-δ-latcone14 undergoes slow hydrolysis to act as a chemical proton source, to assemble DBS-CO2H and (iii) diphenyliodonium nitrate (DPIN)15 is a photo-acid generator to assemble DBS-Gly.
 | ||
| Fig. 1 Gelators used: DBS-CO2H and DBS-Gly (LMWGs) and PEGDM (PG) and triggers HMMP, GdL and DPIN. | ||
Initially, we just tested dual photo-activation of DBS-CO2H and PEGDM. A basic solution of DBS-CO2H was prepared, and to this was added the photo-acid generator DPIN. A mixture of PEGDM and the HMMP photo-initiator was added, and the resulting solution (5 mL) poured into a 5 × 5 cm glass mould (Fig. S1, ESI†). The whole system was cured under high-intensity UV light for 1 h (with ice cooling), and a robust, white, opaque gel formed (Fig. S2, ESI†), presumably as a result of the assembly of both networks (see below). In previous studies,11,13 we observed that PEGDM requires <10 min for crosslinking, while DBS-CO2H needs 60 min for DPIN activation and complete LMWG assembly. We therefore intended to use temporal control to induce differential assembly and gel patterning. The system was initially cured under UV for 10 min, after which a robust, slightly translucent gel had formed, consistent with PEGDM polymerisation. A mask with a series of circles was placed over the gel, and the sample cured for a further 50 min. Opaque circular patterns became visible in the gel (Fig. 2). The opacity results from conversion of DPIN into insoluble iodobenzene, proving significant DPIN activation still occurs after the initial 10 min photoirradiation. This should differentiate the DBS-CO2H network density between patterned and “non-patterned” domains. More sophisticated patterned gels were generated using masks printed on acetate with a laser printer,16 giving good resolution at domain sizes as small as 1 mm (Fig. S3, ESI†).
 | ||
| Fig. 2 Formation of a photopatterned hybrid hydrogel of DBS-CO2H and PEGDM through photoactivation of both networks; after 10 minutes of UV curing a translucent gel is formed mostly based on PEGDM (left); after application of a mask (centre) and 50 minutes of further UV curing, a pattern is visible in the gel as the DPIN is activated and DBS-CO2H self-assembles further (right). | ||
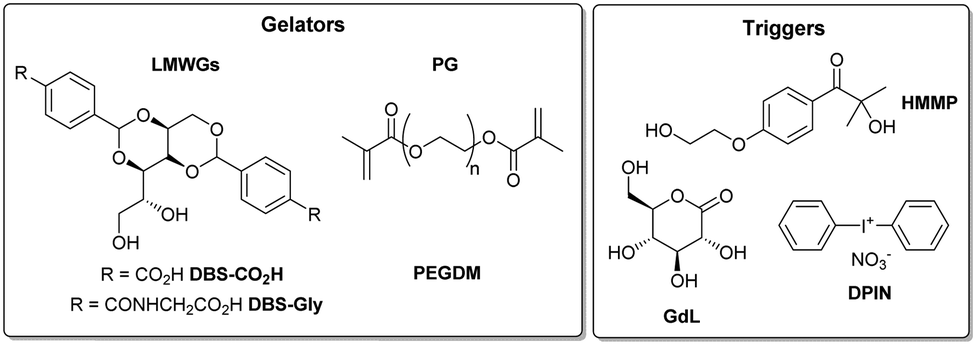
1H NMR studies of DBS-CO2H/PEGDM hybrid hydrogels were performed using DMSO as an internal standard to quantify the extent of self-assembly (mobile gelators are visible in the 1H NMR spectrum prior to assembly and become invisible as they assemble into immobile ‘solid-like’ networks).17 Comparing 1H NMR spectra before and after exposure to UV light for 1 h indicated nearly all of the DBS-CO2H disappeared from the NMR spectrum (Fig. 3), consistent with its UV-driven assembly.
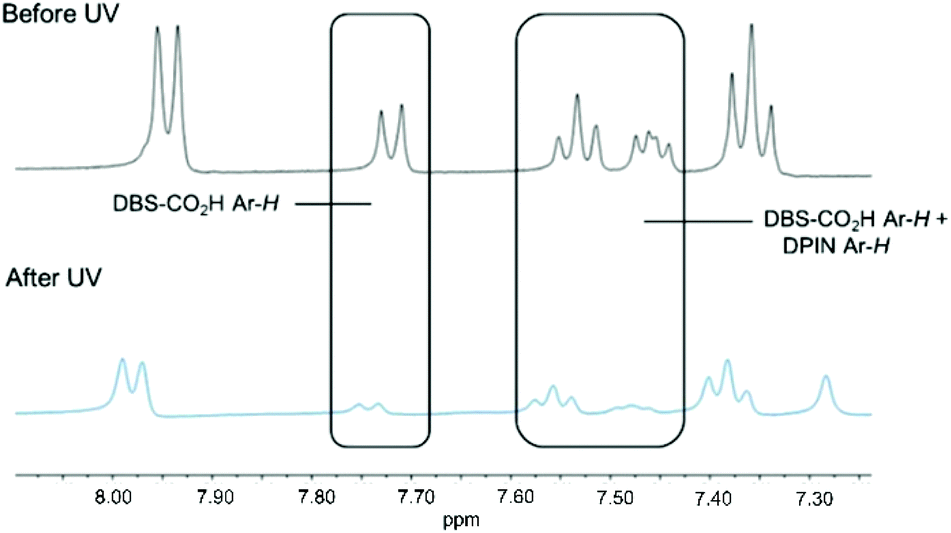 | ||
| Fig. 3 1H NMR of DBS-CO2H (0.4% wt/vol) and PEGDM (5% wt/vol) with DPIN (0.8% wt/vol) as acidifying agent, before and after UV exposure of 1 h. The significant reduction of DBS-CO2H Ar-H peaks signifies that nearly all the LMWG has been protonated. Unlabelled peaks correspond to Ar-H protons of DPIN. | ||
To follow assembly kinetics, multiple NMR samples were made, one being left uncured and the others cured under UV light for different amounts of time. The concentration of mobile DBS-CO2H in each sample was determined (Fig. S4a, ESI†). Photo-induced assembly of DBS-CO2H occurred faster in the presence of PEGDM than in the absence of it (25 min vs. 60 min). We suggest energy transfer from HMMP to DPIN in the multi-component system may increase DPIN hydrolysis, and thus DBS-CO2H assembly. The faster kinetics mean the patterned gels will not be fully self-sorted – both regions will contain some assembled DBS-CO2H, but ‘patterned’ domain will have a greater density. In the absence and presence of PEGDM, DBS-CO2H assembly has an Avrami exponent18 of 1.05 (Fig. S4b, ESI†), suggesting a similar 1-D assembly mechanism in each case.
The hybrid gel was freeze-dried and studied by SEM. A fibrous DBS-CO2H nanostructure was observed, with the fibres appearing embedded/coated in the more film-like nano-structure formed by PEGDM (Fig. S5, ESI†). This would suggest that both PG and LMWG networks are present in the hybrid gel.
Investigations moved on to the assembly of systems that also contained DBS-Gly – three-component gels have rarely been explored. Initially, multi-component hybrid hydrogels of PEGDM, DBS-CO2H and DBS-Gly were prepared using only GdL hydrolysis as a chemical proton source to ascertain whether all three gelators (particularly the two pH-responsive LMWGs) could be sequentially assembled in this complex system. DBS-CO2H and DBS-Gly (each 0.45% wt/vol) were added to PEGDM (5% wt/vol) and HMMP (0.05% wt/vol) in sample vials. The samples were basified with NaOH (0.5 M, aq.) and treated with GdL (18 mg). Samples were cured under UV light for 10 min to obtain a transparent PEGDM hydrogel (Fig. S6, ESI†), which was left overnight for GdL-induced gelation of DBS-CO2H and DBS-Gly. The different pKa values of DBS-CO2H and DBS-Gly11 mean DBS-CO2H should assemble first and DBS-Gly second. The gel changed from clear to translucent (Fig. S5, ESI†), indicative of LMWG assembly – though not indicating the amount of each LMWG in the network, or if they sequentially assemble.
We studied this three-component system further using 1H NMR spectroscopy. Recording spectra immediately after UV curing and after the sample was left for 24 h, demonstrated that nearly all the DBS-CO2H and DBS-Gly were ultimately immobilised (Fig. S7, ESI†). More detailed information was obtained by recording 1H NMR spectra every 30 min after photo-polymerisation, and plotting the concentration of mobile LMWGs (Fig. 4). In the initial 3 h after photo-irradiation, some of each gelator (ca. 25–30%) was immobilised. Gelation then enters a second phase where all of the DBS-CO2H slowly assembles while the concentration of DBS-Gly in solution remains roughly constant. In the third phase, the concentration of DBS-Gly begins to drop more rapidly. There is thus evidence of sequential assembly after some initial immobilisation of both gelators. Experimental limitations meant spectra could only be recorded for 14 h, insufficient for all of the DBS-Gly to assemble, but extrapolating the data, and the 24 h measurement, indicate network assembly is effectively complete by then. This is slower assembly than for the individual gelators or indeed for the two-component LMWG system with a similar amount of GdL in the absence of PEGDM.11 This is probably due to the increase in the viscosity of the solution caused by polymerised PEGDM limiting LMWG diffusion and fibre organisation.19
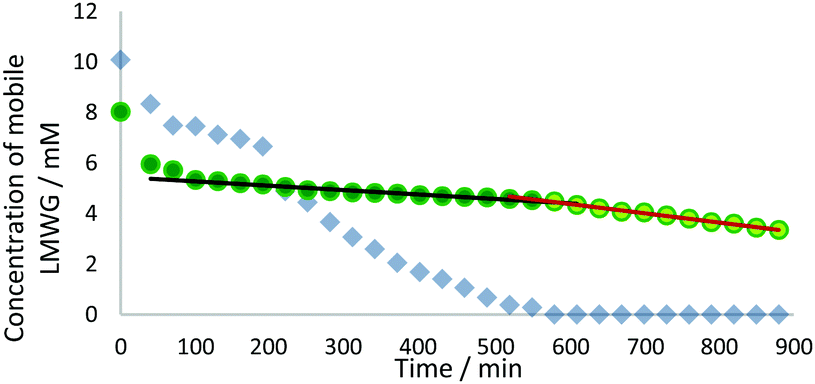 | ||
| Fig. 4 Plot of concentrations of DBS-CO2H (blue diamonds) and DBS-Gly (green circles) (both 0.45% wt/vol) in the presence of PEGDM (5% wt/vol), HMMP (0.05% wt/vol) and GdL (1.8% wt/vol) after irradiation with UV light for 10 min. The different phases of assembly for DBS-Gly are highlighted (dark green circles and black line, light green circles and red line). | ||
Circular dichroism (CD) spectroscopy was performed. After 5 h, an intense CD band at ca. 261 nm, associated with DBS-CO2H, was visible (Fig. S8, ESI†). Unfortunately, the CD band(s) associated with DBS-Gly were obscured by the gluconic acid band. Nonetheless, DBS-CO2H was able to access the expected chiral nanostructure.11,13 SEM imaging of the freeze-dried multi-component hybrid hydrogel showed a LMWG fibre network coated with, or embedded in, PEGDM polymer films (Fig. S9, ESI†). This does not confirm self-sorting of LMWG networks, but shows the gel maintains its hybrid nanoscale morphology. Rheology indicated gel-like behaviour (G′ > G′′). G′ and G′′ for the hybrid hydrogel were greater than for the individual gels (Fig. S10 and S11, ESI†) presumably because the multiple networks reinforce one another. The covalently crosslinked PEGDM network also increased gel stiffness (G′) and resistance to strain (G′/G′′ crossover point), making the gels much more easily handled.
Having ascertained all three gelators could assemble in the same sample, with a degree of control, the next step was to introduce DPIN as a second proton source for greater control. Initially, multi-component hybrid hydrogels with sample-spanning networks of each gelator were prepared (Fig. S12, ESI†). A basified sample of DBS-CO2H and DBS-Gly was mixed with PEGDM, HMMP, enough GdL to theoretically protonate only the DBS-CO2H, and DPIN (a six-component mixture). The solution was poured into a 5 × 5 cm glass mould, and cured under UV light (with cooling) for 10 min to form a robust, slightly translucent PEGDM gel. The mould was left overnight (covered, to prevent drying) for GdL hydrolysis. The gel was then slightly more translucent, indicative of LMWG network assembly (presumably mostly DBS-CO2H, see Fig. 4). This gel was cured under UV for a further 50 min (with cooling) to activate the remaining DPIN. The final gel was more robust, and opaque, indicating DPIN activation and LMWG network assembly (mostly DBS-Gly).
The system was investigated by 1H NMR spectroscopy. It was difficult to quantify how much of each gelator was assembled in the network, due to overlapping aromatic signals from DPIN and signal broadening (Fig. S13, ESI†), but it was possible to qualitatively observe the disappearance of 1H NMR resonances. After the initial, short UV cure to polymerise PEGDM, resonances associated with both LMWGs and DPIN were visible, while those associated with PEGDM (excluding the large polymer CH2 resonance) had disappeared. After leaving for GdL activation, some resonances were significantly reduced, supporting our model in which DBS-CO2H is protonated by GdL. After the second, longer exposure to UV light, the signals corresponding to LMWGs/DPIN decreased further. This shows the LWMGs were assembled in a stepwise manner, and it is likely three networks are present in this integrated gel.
SEM images of the hybrid multi-component gels both before and after UV-activation of DPIN have networks that are similar in appearance, appearing to be coated/embedded in the film-like nanostructure of PEGDM (Fig. S14, ESI†). The gel nanostructure before DPIN activation appears more “globular”, potentially due to less LMWG network being present, hence becoming more thickly coated in the polymer on freeze-drying.
For preliminary investigations of photopatterning, a solution of all three gelators (DBS-CO2H, DBS-Gly and PEGDM), along with the three activating agents HMMP, GdL and DPIN, was poured into a 5 × 5 cm glass mould. A mask obscuring half of the sample was placed over it and the sample cured under UV light for 10 min to polymerise PEGDM, after which the sample was left overnight for GdL hydrolysis. The next day, the mask was rotated 90°, and the sample cured under UV again, for 50 min, to fully activate DPIN in half the sample – this also activates the PI in the previously masked region and cause further formation of the PEGDM gel. A multi-domain multi-component system appeared to be fabricated (Fig. S15, ESI†). However, after GdL hydrolysis (but before the second UV cure), the two domains without a PEGDM network were very fragile. It is possible the presence of so many latent components hinders GdL activation of DBS-CO2H, or DBS-CO2H assembly into fibres, suggesting we are close to the limit with so many components present.
We decided that to improve the mechanical properties of the non-hybrid domains, after the initial UV cure the non-gelled solution should be poured off and replaced by a fresh solution of DBS-CO2H/DBS-Gly with GdL and DPIN (i.e. without PEGDM/HMMP). In this case, the domains in which PEGDM had not been activated were much less fragile. This second solution would sometimes flow a little over the PEGDM gel, so the domains were not so well-defined. Pleasingly, however, we could generate a gel with four visibly distinct domains (Fig. 5):
 | ||
| Fig. 5 Stepwise formation of a four-domain multi-component hybrid hydrogel of DBS-CO2H, DBS-Gly and PEGDM. A solution of all three gelators and HMMP, GdL and DPIN is cured under UV light (with a mask obscuring half the sample) for 10 min to form a PEGDM gel (with a small amount of DBS-CO2H activation). The remaining solution is poured away and replaced by one containing DBS-CO2H, DBS-Gly, GdL and DPIN. The sample is left overnight for GdL to hydrolyse, and partial LMWG assembly (mostly DBS-CO2H). Further UV curing (50 min with mask rotated 90°) activates remaining DPIN and completes LMWG network assembly (mostly DBS-Gly), with iodobenzene by-product making these gel domains opaque. | ||
(1) A domain exposed to short-timescale UV irradiation, GdL, and long-timescale UV, having all three gelator networks assembled in the order PEGDM, DBS-CO2H, DBS-Gly – corresponding to the system characterised in detail above.
(2) A domain exposed only to short timescale UV irradiation and GdL, consisting of a PEGDM network and mostly assembled DBS-CO2H. This system, prior to DBS-Gly assembly, was characterised by 1H NMR above (Fig. 4, t = 600 min).
(3) A domain exposed to no irradiation, and only experiencing GdL activation, consisting mostly of assembled DBS-CO2H and non-assembled DBS-Gly (see ref. 11).
(4) A domain exposed to GdL and long timescale UV irradiation, consisting of assembled DBS-CO2H and DBS-Gly (see ref. 11).
In domains 2 and 3, there will be latent components (e.g. DPIN and DBS-Gly) which could be further activated and/or patterned by additional applications of UV light – work to make use of latent components within such gels is ongoing in our labs.
In summary, we have demonstrated it is possible to co-assemble three different gelators – two LMWGs (DBS-CO2H and DBS-Gly) and a PG (PEGDM). By careful spatial and temporal application of different triggers (physical and chemical) it is possible to sequentially switch-on the assembly of each of these gelators in turn, and in different orders. The gelators are not completely orthogonal, as demonstrated by 1H NMR studies, but there is evidence of sequential LMWG assembly as a result of their different pKa values and some differentiation between photo-responsive gelators based on irradiation time. This system enables photo-patterning of a gel with four different domains depending on the stimuli applied to each part of the sample. The resulting gel forms from a complex mixture of components (three gelators and three triggering agents) in a way that depends on the precise history of each domain. Further work will focus on more detailed characterisation of complex multi-domain gels and use these patterned materials in applications such as tissue engineering,20 where the presence of different domains should lead to differences in the behaviour of stem cells grown on and/or in each domain.
We thank Meg Stark (Dept. of Biology, York) for SEM imaging and University of York for funding.
Conflicts of interest
There are no conflicts to declare.References
- Hydrogels: Recent Advances, ed. V. K. Thakur and M. K. Thakur, Springer, Singapore, 2018 Search PubMed.
- Polymer Gels: Fundamentals and Applications, ed. H. B. Bohidar, P. Dubin and Y. Osada, American Chemical Society, Washington, 2002 Search PubMed.
- (a) R. G. Weiss, J. Am. Chem. Soc., 2014, 136, 7519–7530 CrossRef CAS PubMed; (b) E. R. Draper and D. J. Adams, Chem, 2017, 3, 390–410 CrossRef CAS.
- (a) A. R. Hirst and D. K. Smith, Chem. – Eur. J., 2005, 11, 5496–5508 CrossRef CAS PubMed; (b) L. E. Buerkle and S. J. Rowan, Chem. Soc. Rev., 2012, 41, 6089–6102 RSC; (c) J. Raeburn and D. J. Adams, Chem. Commun., 2015, 51, 5170–5180 RSC; (d) E. R. Draper and D. J. Adams, Chem. Soc. Rev., 2018, 47, 3395–3405 RSC.
- Selected examples: (a) K. Sugiyasu, S.-i. Kawano, N. Fujita and S. Shinkai, Chem. Mater., 2008, 20, 2863–2865 CrossRef CAS; (b) J. R. Moffat and D. K. Smith, Chem. Commun., 2008, 316–318 Search PubMed; (c) A. Bimalendu, J. Nanda and A. Banerjee, Soft Matter, 2011, 7, 8913–8922 RSC; (d) E. R. Draper, J. R. Lee, M. Wallace, F. Jackel, A. J. Cowan and D. J. Adams, Chem. Sci., 2016, 7, 6499–6505 RSC; (e) N. Singh, K. Zhang, C. A. Angulo-Pachon, E. Mendes, J. H. van Esch and B. Escuder, Chem. Sci., 2016, 7, 5568–5572 RSC; (f) S. Onogi, H. Shigemitsu, T. Yoshii, T. Tanida, M. Ikeda, R. Kubota and I. Hamachi, Nat. Chem., 2016, 8, 743–752 CrossRef CAS PubMed; (g) H. Shigemitsu, T. Fujisaku, W. Tanaka, R. Kubota, S. Minami, K. Urayama and I. Hamachi, Nat. Nanotechnol., 2018, 13, 165–172 CrossRef CAS PubMed; (h) B. O. Okesola, Y. Wu, B. Derkus, S. Gani, D. Wu, D. Knani, D. K. Smith, D. J. Adams and A. Mata, Chem. Mater., 2019, 31, 7883–7897 CrossRef CAS PubMed; (i) W. Tanaka, H. Shigemitsu, T. Fujisaku, R. Kubota, S. Minami, K. Urayama and I. Hamachi, J. Am. Chem. Soc., 2019, 141, 4997–5004 CrossRef CAS PubMed.
- D. J. Cornwell and D. K. Smith, Mater. Horiz., 2015, 2, 279–293 RSC.
- P. R. A. Chivers and D. K. Smith, Nat. Rev. Mater., 2019, 4, 463–478 CrossRef CAS.
- (a) J. Eastoe, M. Sánchez-Dominguez, P. Wyatt and R. K. Heenan, Chem. Commun., 2004, 2608–2609 RSC; (b) J. J. D. de Jong, P. R. Hania, A. Pugžlys, L. N. Lucas, M. de Loos, R. M. Kellogg, B. L. Feringa, K. Duppen and J. H. van Esch, Angew. Chem., Int. Ed., 2005, 44, 2373–2376 CrossRef CAS PubMed; (c) C. Maity, W. E. Hendriksen, J. H. van Esch and R. Eelkema, Angew. Chem., Int. Ed., 2015, 54, 998–1001 CrossRef CAS PubMed.
- (a) S. Matsumoto, S. Yamaguchi, S. Ueno, H. Komatsu, M. Ikeda, K. Ishizuka, Y. Iko, K. V. Tabata, H. Aoki, S. Ito, H. Noji and I. Hamachi, Chem. – Eur. J., 2008, 14, 3977–3986 CrossRef CAS PubMed; (b) H. Komatsu, S. Tsukiji, M. Ikeda and I. Hamachi, Chem. – Asian J., 2011, 6, 2368–2375 CrossRef CAS PubMed.
- E. R. Draper, E. G. B. Eden, T. O. McDonald and D. J. Adams, Nat. Chem., 2015, 7, 848–852 CrossRef CAS PubMed.
- D. J. Cornwell, O. J. Daubney and D. K. Smith, J. Am. Chem. Soc., 2015, 137, 15486–15492 CrossRef CAS PubMed.
- (a) X. Che, B. Bai, T. Zhang, C. Zhang, C. Zhang, P. Zhang, H. Wang and M. Li, New J. Chem., 2017, 41, 8614–8619 RSC; (b) P. R. A. Chivers and D. K. Smith, Chem. Sci., 2017, 8, 7218–7227 RSC; (c) C. C. Piras and D. K. Smith, Chem. – Eur. J., 2019, 25, 11318–11326 CAS; (d) L. Thomson, R. Schweins, E. R. Draper and D. J. Adams, Macromol. Rapid Commun., 2020, 2000093 CrossRef CAS PubMed.
- D. J. Cornwell, B. O. Okesola and D. K. Smith, Angew. Chem., Int. Ed., 2014, 53, 12461–12465 CAS.
- D. J. Adams, M. F. Butler, W. J. Frith, M. Kirkland, L. Mullen and P. Sanderson, Soft Matter, 2009, 5, 1856–1862 RSC.
- J. Raeburn, T. O. McDonald and D. J. Adams, Chem. Commun., 2012, 48, 9355–9357 RSC.
- M. S. Hahn, J. S. Miller and J. L. West, Adv. Mater., 2006, 18, 2679–2684 CrossRef CAS.
- (a) B. Escuder, M. Llusar and J. F. Miravet, J. Org. Chem., 2006, 71, 7747–7752 CrossRef CAS PubMed; (b) S. M. Ramalhete, K. P. Nartowski, N. Sarathchandra, J. S. Foster, A. N. Round, J. Angulo, G. O. Lloyd and Y. Z. Khimyak, Chem. – Eur. J., 2017, 23, 8014–8024 CrossRef CAS PubMed.
- (a) M. Avrami, J. Chem. Phys., 1939, 7, 103–112 CrossRef; (b) M. Avrami, J. Chem. Phys., 1940, 8, 212 CrossRef CAS; (c) M. Avrami, J. Chem. Phys., 1941, 9, 177–184 CrossRef CAS.
- G. Pont, L. Chen, D. G. Spiller and D. J. Adams, Soft Matter, 2012, 8, 7797–7802 RSC.
- B. O. Okesola and A. Mata, Chem. Soc. Rev., 2018, 47, 3721–3736 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Materials and methods and additional experimental data. See DOI: 10.1039/d0cc03004j |
| This journal is © The Royal Society of Chemistry 2020 |