
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Gold-linked strings of donor–acceptor dyads: on-surface formation and mutual orientation†
Sujoy
Karan‡
 *a,
Yan
Geng§
b,
Silvio
Decurtins
b,
Shi-Xia
Liu
b and
Jascha
Repp
a
*a,
Yan
Geng§
b,
Silvio
Decurtins
b,
Shi-Xia
Liu
b and
Jascha
Repp
a
aInstitute of Experimental and Applied Physics, University of Regensburg, 93053 Regensburg, Germany. E-mail: sujoy.karan@physik.uni-regensburg.de
bDepartment of Chemistry and Biochemistry, University of Bern, 3012 Bern, Switzerland
First published on 6th June 2020
Abstract
Strings of gold-organic oligomers of polar units have been formed by on-surface synthesis and investigated with non-contact atomic force microscopy. The mutual alignment of dipoles within the strings is analyzed. While an alternating head-to-tail alignment might be expected from dipolar interactions, a more complicated alignment order is observed. The data suggests that coordination bonding to additional gold adatoms leads to stabilization of parallel pairs of molecules, suppressing a head-to-tail alignment order.
Conjugated polymers comprising electron donor and acceptor units exhibit electronic properties suitable for applications in advanced organic devices such as field effect transistors, light emitting diodes, and photovoltaics.1–6 An efficient electron transfer along these polymers depends critically on the relative alignment of monomer units. Great progress in the synthesis of molecular networks on surfaces has been achieved,7–9 demonstrating novel possibilities to steer the pattern formation by various means. Recently, surface chemistry has been exploited to engineer the band structures of polymers synthesized from chemically designed precursors.10,11 In particular, electronic states associated to the topology of the polymers have been at the focus. The controlled implementation of large electric dipoles in such structures, as well as ways to control the mutual alignment of neighboring units would add a novel aspect to the toolset of band-structure engineering. However, polymers possessing definite arrangements of donor and acceptor units have rarely been synthesized via an on-surface synthesis.4 Different approaches have been explored to provide specificity and directionality in molecular self-assembly via weak non-covalent interactions such as hydrogen bonding,12,13 electrostatic interactions,14–17 and metal–ligand coordination.18–21
We synthesize a precursor by annulation of an electron donor and an electron acceptor,3,22,23 leading to an intrinsic dipole moment of ∼5 Debye23 along the molecular axis that is expected to steer the relative alignment of adjacent monomers. The low-energy configuration of dipoles would suggest an antiparallel head-to-tail ordering of monomers.24 Here we demonstrate on-surface synthesis of gold-organic hybrids comprising highly polarizable tetrathiafulvalene–benzothiadiazole (TTF–BTD) dyads exhibiting a complicated short-range order that is lost within a few molecular units. The parallel and antiparallel arrangements of neighboring monomers appear to be correlated with the existence or absence of additional gold coordination bonds, as identified directly by means of non-contact atomic force microscopy (nc-AFM) with sub-angstrom lateral resolution. Formation of aryl–aryl bonds between molecules is not observed, presumably owing to Coulomb repulsion between lone-pair electrons.
Dibromo-TTF–BTD dyads (inset of Fig. 1a) were synthesized (see details in ESI†) via a reaction of 4,8-dibromo[1,3]dithiolo[4,5-f]-2,1,3-benzothiadiazol-6-one with vinylene trithiocarbonate in presence of triethylphosphite in 35% yield. The molecules were sublimated onto Au(111) substrates at temperatures below ∼12 K and later annealed at elevated temperatures to induce debromination. Prior to annealing, we investigated the surface to exclude dissociation of molecules upon deposition, and ensured the purity of the precursors. Experiments were performed with a qPlus-based25 combined scanning tunneling and atomic force microscope operated at ∼9 K under ultra-high vacuum. A PtIr wire was used as the tip, treated in situ by indenting into the substrate. CO-terminated tips were used for acquiring frequency shift (Δf) images.26
 | ||
| Fig. 1 (a) Large-scale STM image shows strings of molecules on Au(111). A ball and stick model of the precursor molecule (dibromo-TTF–BTD) is shown as an inset, where grey, blue, yellow, and red represent carbon, nitrogen, sulphur, and bromine, respectively. The π-conjugated molecular backbone comprises a tetrathiafulvalene (TTF) and a 2,1,3-benzothiadiazole (BTD) moiety fused in a nearly-planar geometry.24 TTF–BTD possesses an in-plane dipole as it consists of a strong electron donor (TTF)27 and an electron acceptor (BTD). (b) Zoom-in of a chain segment, acquired with a metal tip at a sample voltage V = 0.1 V. Scale bars indicate 20 nm in (a) and 1 nm in (b). | ||
Fig. 1a shows chains of TTF–BTD molecules formed upon annealing on Au(111) at 490 K. The chains run preferentially along fcc regions of herringbone reconstructions. Single molecules are observed to be trapped at the reactive elbow sites of the reconstructed surface. Already at first glance, the chains seem to exhibit some short range order, but also pronounced irregularities. Closer inspections (Fig. 1b) using scanning tunneling microscopy (STM) show features related presumably to monomer units, but the detailed internal structure remains elusive.
To discern the atomistic details, the chains were investigated with nc-AFM using a CO-terminated tip26 which enables direct visualization of the chemical structures of surface-supported adsorbates. Fig. 2a shows frequency shift (Δf) image of a chain acquired at a constant tip height. Parallel pairs of molecules are readily identified that are bonded in an ordered array. Inter-monomer bonds are observed where they were expected, namely at the sites that can be thermally activated by debromination. The observed center-to-center spacing of ∼0.72 ± 0.03 nm between adjacent molecules (denoted L in Fig. 2a and b) is too large to be compatible with a covalent aryl–aryl (for which L would have been ∼0.43 ± 0.02 nm) coupling that was expected in analogy to other on-surface Ullmann coupling reaction.28–30
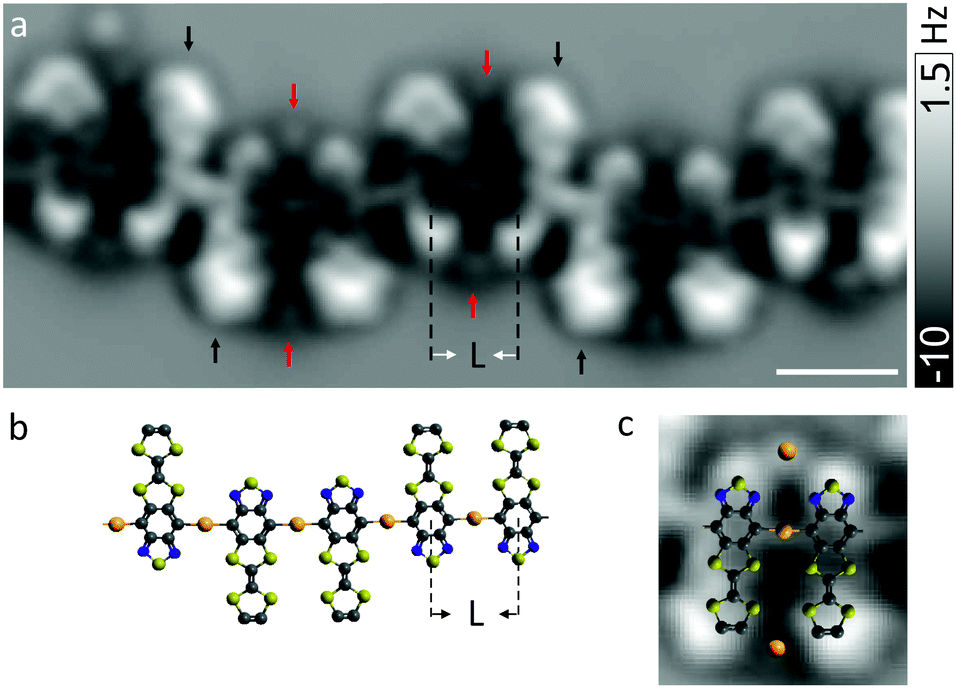 | ||
| Fig. 2 (a) Constant-height Δf image recorded at Δz = 0.16 nm with oscillation amplitude A = 0.05 nm. Zero displacement (Δz = 0) corresponds to I = 1.5 pA and V = 0.2 V above the clean Au surface. Scale bar is indicative of a length of 1 nm. (b) The model of a chain comprising bidentate aryl–gold bonding. Gold atoms are depicted in orange. L denotes the spacing between two nearest neighbors. (c) A model of a parallel pair with two additional gold adatoms overlaid on the frequency-shift image. | ||
Instead, it suggests formation of linear aryl–gold–aryl bonding (i.e., C–Au–C, Fig. 2b) with the separation between two neighboring aryl rings being consistent with previous reports.31–34 It is expected that surface atoms diffuse in large numbers, likely with enhanced mobility in the presence of bromines,35 at temperature to which the sample is annealed, which eventually leads to the formation of organometallic strings. The structural reordering of herringbone reconstruction caused by the adsorption of molecules may act also to release gold adatoms.36–39 A direct coupling between carbon atoms through reductive elimination is presumably hindered owing to Coulomb repulsion between lone pairs of electrons among neighboring monomers. We note that reports on direct observation and geometric characterization of stable gold-organic hybrids31,39,40 are scarce due to relatively low activation barrier of aryl–aryl bond formation between chemisorbed aryl radicals.40
The Δf signal in the AFM image is inhomogeneous across some of the molecules (indicated with black arrows in Fig. 2a), suggesting tilt of molecules with respect to the surface plane. It may arise from a repulsion among lone pairs of electrons on the nitrogen and sulfur atoms of identical neighboring moieties. Moreover, an incommensurability with the substrate lattice may explain why the tilt of molecules is not homogeneous. The influence of surface corrugation due to herringbone reconstruction may be excluded since segments of the chains lain within fcc regions of the reconstructed surface were investigated.
Between pairs of monomers oriented in parallel, small features of repulsive contrast are observed (indicated with red arrows). Such a feature is absent between molecules with antiparallel alignment. We speculate that these features could originate from gold atoms that may be trapped between molecules via coordination bonds41–43 involving nitrogen and sulfur atoms (Fig. 2c). This would straightforwardly explain why these features are only observed between pairs of parallel monomers. Otherwise, one would not expect to observe a stable pair of TTF–BTD molecules arranged in a parallel head-to-head fashion because Coulomb repulsion between equal moieties should favor a head-to-tail arrangement.24 Bending of the CO molecule at the tip apex causes image distortions44,45 and thereby prevents a direct accurate measurement of bond distances. However, the positions of centers of aromatic rings can be determined relatively precisely, such that the bond distances could be extracted by making use of the known geometry of the precursor molecules. Thereby, we estimate the distance between observed additional features and the closest atomic sites of the molecules to ∼0.35 ± 0.07 nm. This seems rather long for a coordinative bond, but appears still compatible with a stabilization mechanism via (weak) coordination bonds.41 Note that the bond lengths may vary with different coordination geometries and the adatoms involved in the coordination may not always exhibit a clear Δf contrast.20,46 We do not exclude the possibility of bromine or carbon monoxide being responsible for or involved in the additional coordination.
We analyze segments of different chains, two of which are presented in Fig. 3a and b showing aperiodic alignments of molecules. While parallel pairs are still seen as majority, segments with more than two neighboring molecules in the same orientation or adjacent antiparallel orientations are also observed. An insight into the order of molecular orientations is provided by an autocorrelation analysis (Fig. 3c) extracted from the segments of different chains containing in total of 166 monomer units. The gray area encompasses values within the standard deviation (≈0.08). The nearest and the next-nearest neighbors appear to be significantly anti-correlated, while every third (and, only very weakly, every sixth) neighbor is found to be in weak correlation. The anticorrelation of next-nearest neighbors reflects a higher probability of finding molecules in pairs (e.g., Fig. 2a). To better comprehend the degree of correlation, a hypothetical autocorrelation for an infinite chain of perfect pairwise ordering of the type -up-up-down-down-up-up-down-down- is shown as an inset of Fig. 3c, which exhibits no nearest-neighbor correlation (because one of the neighbors is always parallel and the other antiparallel) and a perfect anticorrelation of −1 for the next-nearest neighbors (because all next-nearest neighbors are antiparallel). The experimentally observed autocorrelation is far from such a perfect order.
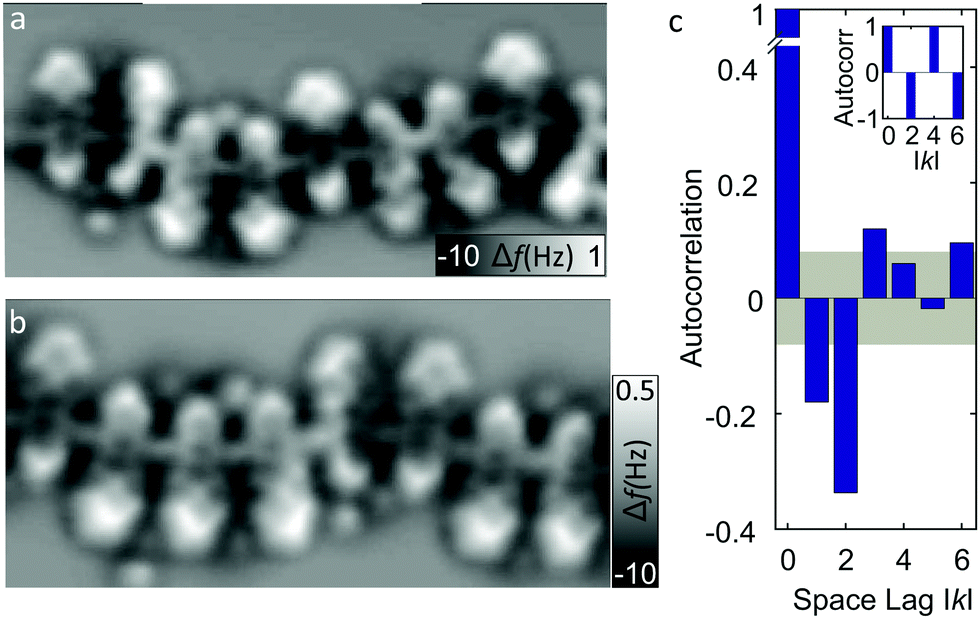 | ||
Fig. 3 (a) and (b) Constant-height Δf images of segments of two different chains (at Δz = 0.18 and 0.16 nm, respectively with A = 0.05 nm) showing irregularities in relative molecular orientations. (c) Autocorrelation analysis of n = 166 molecules from different chains. Gray area marks the standard deviation 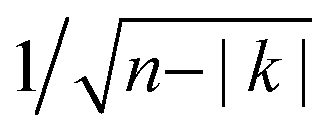 , only values larger than that may be considered significant. In the inset a hypothetical autocorrelation for an infinite chain of perfect pairwise ordering (u-u-d-d-u-u-d-d-) is presented for comparison. , only values larger than that may be considered significant. In the inset a hypothetical autocorrelation for an infinite chain of perfect pairwise ordering (u-u-d-d-u-u-d-d-) is presented for comparison. | ||
In a very simple model, one could assume that the mutual alignment of two adjacent units is given by a certain probability p for parallel and a probability q = 1 − p for antiparallel alignment. This model implies that the probability of parallel or antiparallel alignment is only governed by local interactions between the nearest neighbors. We tested this assumption and observed that this simple model cannot capture the observed autocorrelation irrespective of the value of the parameter (see details in ESI†). Instead assuming that the alignment of the next unit does not only depend on the direct neighbor, but also on the second-nearest neighbor, does qualitatively reproduce the autocorrelation trace (Fig. S1b in ESI†). From this we conclude that the alignment of molecules observed in the experiment must be partially driven by next-nearest-neighbor effects. The slightly canted azimuthal orientation of two neighboring molecules with the same orientation hints towards a locally developed compressive strain in the polymeric chain. The release of such strain may be the driving force for the observed next-nearest-neighbor correlation effects. We note that the number of additional atoms that can be coordinated between each two adjacent monomers depends on their mutual alignment. This mechanism will therefore contribute to the nearest-neighbor correlation effects. However, it is confined to the interaction of direct neighbors and can therefore not explain the observed next-nearest-neighbor correlation effects.
Finally, to address the role of the substrate, experiments were repeated on a more reactive Cu(111) surface. The substrate with sub-monolayer coverages of precursor molecules was annealed to 315 K to facilitate catalytic reaction,47 resulting in dehalogenation and subsequent polymerization of molecular radicals (Fig. 4). Aperiodic 1D networks are observed and bromine atoms, dissociated from the precursor monomers, are found either in close-packed islands or to get trapped at the periphery of the networks. Further annealing of the sample to 385 K did not give rise to appreciable changes in the structures. These results suggest that, indeed, a less reactive surface like gold is more likely to allow for ordered arrangements in on-surface structures, in agreement with previous work.48
 | ||
| Fig. 4 Structures of TTF–BTD assemblies on a Cu(111) substrate (V = 5 mV, I = 15 pA). Bromines dissociated from precursors form islands. Scale bar indicates 5 nm. | ||
In conclusion, 1D strings comprising fused donor–acceptor dyads were prepared by on-surface synthesis on Au(111), with the prospect of a controlled implementation of electric dipoles in surface-supported polymers. The molecules are observed to couple via aryl–gold bonds into hybrid wires, while coordination with additional adatoms inhibits the long-range order in the mutual alignment of adjacent units. A direct aryl–aryl coupling reaction has not occurred, probably owing to Coulomb repulsion between lone pairs of electrons among neighboring monomers. While dipole–dipole interactions would favor an antiparallel orientation of neighboring units, the experimentally observed alignment of molecular dipoles in the chains shows a weak local correlation but no long-range order. We hope that our study will trigger new strategies to facilitate mutual alignment of adjacent units in on-surface synthesis from non-symmetric monomer units in the future.
Financial support from the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation), Project IDs SFB 689 and 314695032 SFB 1277 – B01 is gratefully acknowledged. Open Access funding is provided by the Max Planck Society.
Conflicts of interest
There are no conflicts to declare.Notes and references
- T. A. Zeidan, Q. Wang, T. Fiebig and F. D. Lewis, J. Am. Chem. Soc., 2007, 129, 9848 CrossRef CAS PubMed.
- S. H. Choi and C. D. Frisbie, J. Am. Chem. Soc., 2010, 132, 16191 CrossRef CAS PubMed.
- J. J. Bergkamp, S. Decurtins and S.-X. Liu, Chem. Soc. Rev., 2015, 44, 863 RSC.
- C. Nacci, F. Ample, D. Bleger, S. Hecht, C. Joachim and L. Grill, Nat. Commun., 2015, 6, 7397 CrossRef PubMed.
- A. Amacher, C. Yi, J. Yang, M. P. Bircher, Y. Fu, M. Cascella, M. Graetzel, S. Decurtins and S.-X. Liu, Chem. Commun., 2014, 50, 6540 RSC.
- M. Gilbert and B. Albinsson, Chem. Soc. Rev., 2015, 44, 845 RSC.
- P. A. Held, H. Fuchs and A. Studer, Chem. – Eur. J., 2017, 23, 5874 CrossRef CAS PubMed.
- R. Lindner and A. Kühnle, ChemPhysChem, 2015, 16, 1582 CrossRef CAS PubMed.
- L. Dong, P. N. Liu and N. Lin, Acc. Chem. Res., 2015, 48, 2765 CrossRef CAS PubMed.
- D. J. Rizzo, G. Veber, T. Cao, C. Bronner, T. Chen, F. Zhao, H. Rodriguez, S. G. Louie, M. F. Crommie and F. R. Fischer, Nature, 2018, 560, 204 CrossRef CAS PubMed.
- O. Gröning, S. Wang, X. Yao, C. A. Pignedoli, G. B. Barin, C. Daniels, A. Cupo, V. Meunier, X. Feng, A. Narita, K. Müllen, P. Ruffieux and R. Fasel, Nature, 2018, 560, 209 CrossRef PubMed.
- J. V. Barth, J. Weckesser, C. Cai, P. Günter, L. Bürgi, O. Jeandupeux and K. Kern, Angew. Chem., Int. Ed., 2000, 39, 1230 CrossRef CAS PubMed.
- S. Karan, Y. Wang, R. Robles, N. Lorente and R. Berndt, J. Am. Chem. Soc., 2013, 135, 14004 CrossRef CAS PubMed.
- Y. Wei, W. Tong, C. Wise, X. Wei, K. Armbrust and M. Zimmt, J. Am. Chem. Soc., 2006, 128, 13362 CrossRef CAS PubMed.
- A. E. Baber, S. C. Jensen and E. C. H. Sykes, J. Am. Chem. Soc., 2007, 129, 6368 CrossRef CAS PubMed.
- D. A. Kunkel, S. Simpson, J. Nitz, G. A. Rojas, E. Zurek, L. Routaboul, B. Doudin, P. Braunstein, P. A. Dowben and A. Enders, Chem. Commun., 2012, 48, 7143 RSC.
- L. Xu, X. Miao, B. Zha, K. Miao and W. Deng, J. Phys. Chem. C, 2013, 117, 12707 CrossRef CAS.
- P. F. Cafe, A. G. Larsen, W. Yang, A. Bilic, I. M. Blake, M. J. Crossley, J. Zhang, H. Wackerbarth, J. Ulstrup and J. R. Reimers, J. Phys. Chem. C, 2007, 111, 17285 CrossRef CAS.
- Z. Shi and N. Lin, J. Am. Chem. Soc., 2009, 131, 5376 CrossRef CAS PubMed.
- N. Kocić, X. Liu, S. Chen, S. Decurtins, S. Krejčí, P. Jelínek, J. Repp and S.-X. Liu, J. Am. Chem. Soc., 2016, 138, 5585 CrossRef PubMed.
- L. Dong, Z. Gao and N. Lin, Prog. Surf. Sci., 2016, 91, 101 CrossRef CAS.
- F. Pop, A. Amacher, N. Avarvari, J. Ding, L. M. L. Daku, A. Hauser, M. Koch, J. Hauser, S.-X. Liu and S. Decurtins, Chem. – Eur. J., 2013, 19, 2504 CrossRef CAS PubMed.
- A. Amacher, H. Luo, Z. Liu, M. Bircher, M. Cascella, J. Hauser, S. Decurtins, D. Zhang and S.-X. Liu, RSC Adv., 2014, 4, 2873 RSC.
- Y. Geng, R. Pfattner, A. Campos, J. Hauser, V. Laukhin, J. Puigdollers, J. Veciana, M. Mas-Torrent, C. Rovira, S. Decurtins and S.-X. Liu, Chem. – Eur. J., 2014, 20, 7136 CrossRef CAS PubMed.
- F. J. Giessibl, Appl. Phys. Lett., 2000, 76, 1470 CrossRef CAS.
- L. Gross, F. Mohn, N. Moll, P. Liljeroth and G. Meyer, Science, 2009, 325, 1110 CrossRef CAS PubMed.
- S.-X. Liu, S. Dolder, E. B. Rusanov, H. Stoeckli-Evans and S. Decurtins, C. R. Chim., 2003, 6, 657 CrossRef CAS.
- L. Grill, M. Dyer, L. Lafferentz, M. Persson, M. V. Peters and S. Hecht, Nat. Nanotechnol., 2007, 2, 687 CrossRef CAS PubMed.
- J. Cai, P. Ruffieux, R. Jaafar, M. Bieri, T. Braun, S. Blankenburg, M. Muoth, A. P. Seitsonen, M. Saleh, X. Feng, K. Müllen and R. Fasel, Nature, 2010, 466, 470 CrossRef CAS PubMed.
- Q. Fan, C. Wang, Y. Han, J. Zhu, W. Hieringer, J. Kuttner, G. Hilt and J. M. Gottfried, Angew. Chem., Int. Ed., 2013, 52, 4668 CrossRef CAS PubMed.
- H. Zhang, J.-H. Franke, D. Zhong, Y. Li, A. Timmer, O. D. Arado, H. Mönig, H. Wang, L. Chi, Z. Wang, K. Müllen and H. Fuchs, Small, 2014, 10, 1361 CrossRef CAS.
- H. Zhang, H. Lin, K. Sun, L. Chen, Y. Zagranyarski, N. Aghdassi, S. Duhm, Q. Li, D. Zhong, Y. Li, K. Müllen, H. Fuchs and L. Chi, J. Am. Chem. Soc., 2015, 137, 4022 CrossRef CAS PubMed.
- J. I. Urgel, H. Hayashi, M. D. Giovannantonio, C. A. Pignedoli, S. Mishra, O. Deniz, M. Yamashita, T. Dienel, P. Ruffieux, H. Yamada and R. Fasel, J. Am. Chem. Soc., 2017, 139, 11658 CrossRef CAS PubMed.
- M. Koch, M. Gille, A. Viertel, S. Hecht and L. Grill, Surf. Sci., 2014, 627, 70 CrossRef CAS.
- L. Huang, P. Zeppenfeld, S. Horch and G. Comsa, J. Chem. Phys., 1997, 107, 585 CrossRef CAS.
- T. A. Baker, E. Kaxiras and C. M. Friend, Top. Catal., 2010, 53, 365 CrossRef CAS.
- S. M. Driver, T. Zhang and D. A. King, Angew. Chem., Int. Ed., 2007, 46, 700 CrossRef CAS PubMed.
- B. K. Min, X. Deng, D. Pinnaduwage, R. Schalek and C. M. Friend, Phys. Rev. B: Condens. Matter Mater. Phys., 2005, 72, 121410 (R) CrossRef.
- A. Saywell, W. Greń, G. Franc, A. Gourdon, X. Bouju and L. Grill, J. Phys. Chem. C, 2014, 118, 1719 CrossRef CAS.
- H. Zhang and L. Chi, Adv. Mater., 2016, 28, 10492 CrossRef CAS PubMed.
- R. Pawlak, T. Meier, N. Renaud, M. Kisiel, A. Hinaut, T. Glatzel, D. Sordes, C. Durand, W.-H. Soe, A. Baratoff, C. Joachim, C. E. Housecroft, E. C. Constable and E. Meyer, ACS Nano, 2017, 11, 9930 CrossRef CAS PubMed.
- Z. Feng, S. Velari, A. Cossaro, C. Castellarin-Cudia, A. Verdini, E. Vesselli, C. Dri, M. Peressi, A. De Vita and G. Comelli, ACS Nano, 2015, 9, 8697 CrossRef CAS PubMed.
- S.-W. Hla, ACS Nano, 2015, 9, 8644 CrossRef CAS PubMed.
- M. Neu, N. Moll, L. Gross, G. Meyer, F. J. Giessibl and J. Repp, Phys. Rev. B: Condens. Matter Mater. Phys., 2014, 89, 205407 CrossRef.
- N. Moll, B. Schuler, S. Kawai, F. Xu, L. Peng, A. Orita, J. Otera, A. Curioni, M. Neu, J. Repp, G. Meyer and L. Gross, Nano Lett., 2014, 14, 6127 CrossRef CAS PubMed.
- S. Zint, D. Ebeling, D. Schlöder, S. Ahles, D. Mollenhauer, H. A. Wegner and A. Schirmeisen, ACS Nano, 2017, 11, 4183 CrossRef CAS PubMed.
- H. Walch, R. Gutzler, T. Sirtl, G. Eder and M. Lackinger, J. Phys. Chem. C, 2010, 114, 12604 CrossRef CAS.
- R. Gutzler, H. Walch, G. Eder, S. Kloft, W. M. Heckl and M. Lackinger, Chem. Commun., 2009, 4456 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available: Synthesis of dibromo-TTF-BTD, autocorrelation of simulated chains. See DOI: 10.1039/d0cc02990d |
| ‡ Present address: Max Planck Institute for Solid State Research, Heisenbergstraße 1, 70569 Stuttgart, Germany. E-mail: s.karan@fkf.mpg.de |
| § Present address: College of Chemistry, Chemical Engineering and Material Science, Shandong Normal University, Jinan 250014, P. R. China. |
| This journal is © The Royal Society of Chemistry 2020 |