
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDetecting protein–protein interactions by Xe-129 NMR†
Zhuangyu
Zhao
 ,
Benjamin W.
Roose
,
Serge D.
Zemerov
,
Madison A.
Stringer
and
Ivan J.
Dmochowski
*
,
Benjamin W.
Roose
,
Serge D.
Zemerov
,
Madison A.
Stringer
and
Ivan J.
Dmochowski
*
Department of Chemistry, University of Pennsylvania, Philadelphia, Pennsylvania 19104, USA. E-mail: ivandmo@sas.upenn.edu
First published on 20th August 2020
Abstract
Detection of protein–protein interactions (PPIs) is limited by current bioanalytical methods. A protein complementation assay (PCA), split TEM-1 β-lactamase, interacts with xenon at the interface of the TEM-1 fragments. Reconstitution of TEM-1—promoted here by cFos/cJun leucine zipper interaction—gives rise to sensitive 129Xe NMR signal in bacterial cells.
The analysis of protein–protein interactions (PPIs) is crucial for understanding protein functions and cell physiology. The protein complementation assay (PCA) is widely used for in vivo detection and validation of PPIs, whereby two catalytically or spectroscopically inactive halves of a reporter protein, e.g., β-lactamase, green fluorescent protein (GFP), or luciferase, are fused to a pair of potentially interacting proteins.1–9 The PPI reconstitutes the functional reporter, which gives a detectable readout such as enzymatic activity, fluorescence, or bioluminescence.3–5,10–12 While enzymatic activity can be readily monitored by cell resistance to addition of antibiotics or deprivation of nutrient, and is thus useful for high-throughput screening, it is less suitable for obtaining spatiotemporal information. Previous studies mostly relied on fluorescence and luminescence to probe dynamic PPIs.13–20 These imaging techniques are successfully applied to biochemical studies in cell lines and small, transparent model organisms.5,6,14,21 However, optical approaches have limitations introduced by nonspecific protein binding, endogenous fluorescence quenchers, and varying intracellular environments, such as pH and oxygenation, which can affect quantum yield and rates of photobleaching. New analytical methods for studying PPIs are needed, particularly to enable in vivo studies.
Nuclear magnetic resonance (NMR) spectroscopic techniques have evolved as useful tools to monitor PPIs that are difficult to study by other methods, and provide structural details of the protein interfaces.22–30 However, NMR experiments using 1H, 13C and 15N nuclei often suffer from low sensitivity, especially for medium to large protein systems, poor selectivity due to complicated biological environments, and usually require isotopic labelling. 129Xe NMR can provide good selectivity due to low intrinsic background signal and excellent detection sensitivity owing to hyperpolarized (hp) 129Xe chemical exchange saturation transfer (hyper-CEST).31 This NMR technique applies selective saturation pulses at the 129Xe-host chemical shift and monitors depolarization of 129Xe nuclear spins in aqueous solution. Hyper-CEST enables nM-to-fM detection of molecules capable of transiently encapsulating xenon.32–41 Herein, we report TEM-1 β-lactamase as a 129Xe NMR-based PCA reporter, which gives “turn-on” hyper-CEST signal upon assembly of the TEM-1 fragments, T1Fα and T1Fω. TEM-1 assembly was driven by the well-characterized cFos/cJun leucine zipper PPI,42 which was determined to be readily detectable in bacterial cells.
TEM-1 β-lactamase has been one of the most used PCA reporters since the identification of its α and ω fragments.2,3 The enzymatic activity of TEM-1 can be used as a proxy to monitor the interaction of the two fusion proteins.2,43 In some studies, enzymatic activity can be monitored by the conversion of substrate into colorimetric or fluorescent products.13,44 Moreover, our laboratory previously found that TEM-1 can function as a genetically encoded reporter for 129Xe hyper-CEST NMR.45 Notably, 129Xe exchanging between TEM-1 and aqueous solvent gives rise to a hyper-CEST signal at +60 ppm, referenced to the 129Xe@aq signal. X-ray crystallography, molecular dynamics (MD) simulations, and protein mutagenesis established the Xe binding site responsible for generating CEST contrast from wild-type TEM-1, which coincidentally resides at the interface between the α and ω fragments of split TEM-1 (Fig. 1).45,46 We therefore ventured that CEST contrast could be used to monitor PPIs in a turn-on manner using TEM-1 fragment complementation. To investigate this method, we used the α and ω TEM-1 fragments employed by Wehrman et al.3 and fused them to complementary cFos and cJun sequences, respectively (T1Fα-cFos and cJun-T1Fω; Scheme 1). Purity and secondary structures of TEM-1 fragments were confirmed by gel electrophoresis and circular dichroism spectroscopy, respectively (Fig. S1 and S2, ESI†). All protein samples remained well-dispersed (non-aggregated) before and after hyper-CEST experiments, as confirmed by dynamic light scattering (Fig. S3, ESI†).
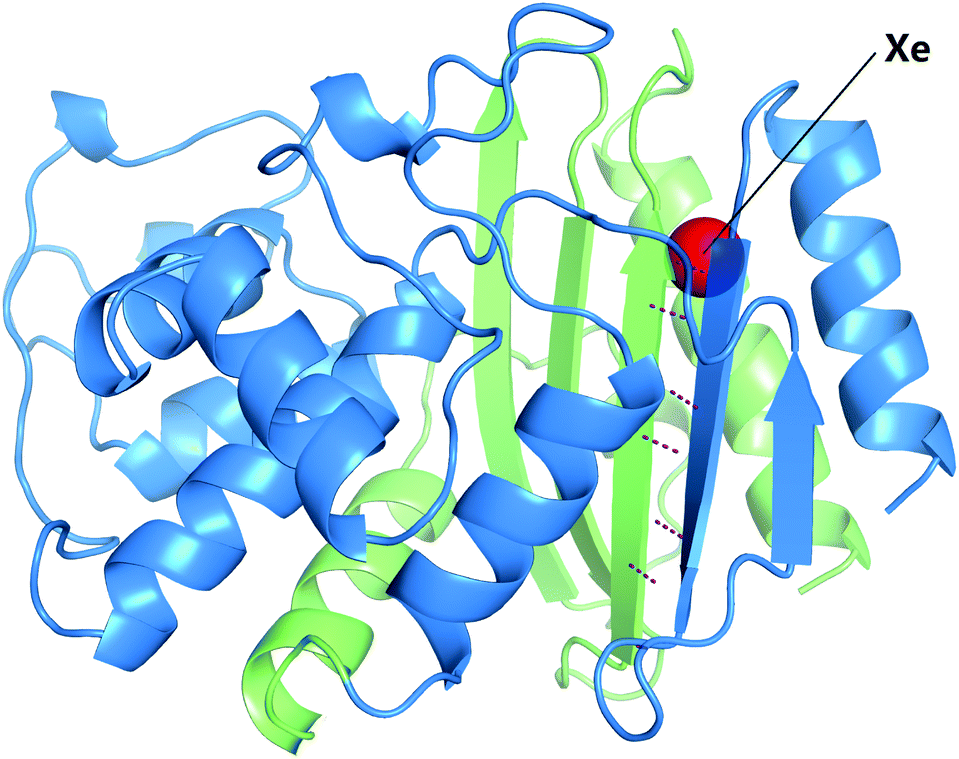 | ||
| Fig. 1 T1Fα (blue; residues 26–197) and T1Fω fragments (green; residues 198–290) indicated for the crystal structure of TEM-1 derivatized with Xe (PDB ID 5HW1). For simplicity, only the primary xenon site is shown (red atom). Hydrogen bonds between the backbones of the two β-strands at the interface are shown as purple dashed lines. | ||
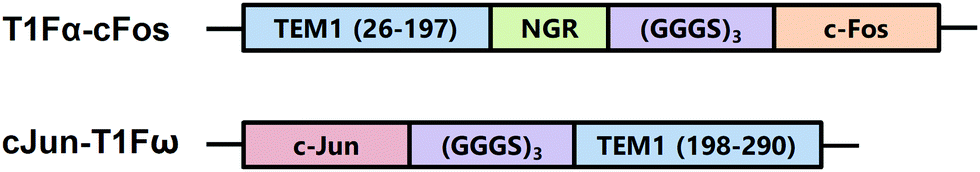 | ||
| Scheme 1 TEM-1 β-lactamase fragment constructs used in this study. | ||
Hyper-CEST z-spectra of 80 μM purified recombinant T1Fα-cFos and cJun-T1Fω were obtained using multiple selective d-SNOB saturation pulses (Fig. 2). In the presence of only one protein fragment, no 129Xe@protein signal was observable, indicating lack of a site compatible with Xe exchange. In contrast, a sample containing both fragments at equimolar concentration showed a saturation peak at +56 ppm, very close to the chemical shift of 129Xe@wtTEM-1. This suggested that hetero-dimerization of T1Fα-cFos/cJun-T1Fω resembled the protein structure and Xe binding site of wild-type TEM-1. The small difference in chemical shift is likely modulated by slight changes to the protein dynamics and by the presence of cFos/cJun. Notably, the 129Xe@T1Fα-cFos/cJun-T1Fω signal arose from assembly of the two fragments, thereby excluding non-specific protein binding that sometimes can give false signal in enzymatic activity or spectrophotometric assays. The magnitude of the CEST effect (1 − Mz/M0 = 0.22) was attenuated compared to wild-type TEM-1 (1 − Mz/M0 = 0.62),45 which is likely due to faster Xe exchange and weaker Xe binding affinity as a result of greater protein dynamics and altered topology of split TEM-1. Homo-dimerization of cFos and cJun proteins may also play a role at high micromolar concentrations.47,48
 | ||
| Fig. 2 Hyper-CEST z-spectra of 80 μM T1Fα-cFos, 80 μM cJun-T1Fω and 80 μM both fragments in 50 mM Tris (pH 7.4), 500 mM NaCl at 300 K. Saturation time, tsat = 2.29 s; field strength, B1,max = 77 μT. The solid circles show the experimental data, and the lines show the Lorentzian fits. Each spectrum is the average of three measurements. | ||
To evaluate the detection sensitivity, time-dependent saturation transfer experiments were performed by measuring the 129Xe(aq) signal against varying saturation time using saturation pulses centered at frequency offsets of +56 ppm and −56 ppm, for on- and off-resonance, respectively. T1Fα-cFos/cJun-T1Fω complex (1 μM) gave rise to 0.06 ± 0.01 saturation contrast (Fig. S4, ESI†). To verify that the observed contrast was dependent on the interaction of cFos and cJun, the T1Fα fragment was prepared without the C-terminal cFos and mixed with equimolar cJun-T1Fω fragment. As expected, 1 μM T1Fα/cJun-T1Fω showed minimal background signal with saturation contrast of 0.01 ± 0.01 (Fig. S4, ESI†). This indicated that the limit of detection (LOD) for cFos/cJun PPI was roughly 1 μM. Given that the binding affinity between cFos and cJun is at high nanomolar level,48 the signal-to-noise ratio and LOD should improve somewhat for stronger protein–protein interactions. The result also indicates that this TEM-1-based approach is useful to probe PPIs with medium to strong affinity (dissociation constant, Kd < 1 μM). For weak PPIs (Kd > 10 μM), reporter concentration higher than Kd is required for detection.
To investigate the possibility that weak interaction between T1Fα and T1Fω may contribute to background signal at high concentrations, we carried out hyper-CEST experiments with the cFos deletion construct at a concentration of 80 μM. A z-spectrum of 80 μM T1Fα/cJun-T1Fω using 77 μT saturation pulses showed a saturation response at +48 ppm somewhat similar to that of T1Fα-cFos/cJun-T1Fω, indicating reconstitution of TEM-1 (Fig. S5, ESI†). This probably results from increased thermostability due to several hydrogen bonds between β-strands at the interface of the two fragments and a reduction of hydrophobic surface area (Fig. 1). Meanwhile, cFos deletion caused a broader peak width of the 129Xe NMR signal, implying faster 129Xe exchange and greater protein dynamics. Time-dependent saturation transfer experiments using 279 μT saturation pulses confirmed that 80 μM T1Fα/cJun-T1Fω gave rise to the same saturation contrast as T1Fα-cFos/cJun-T1Fω (0.44 ± 0.02 and 0.42 ± 0.02, respectively; Fig. S6, ESI†). As non-specific Xe–protein interactions sometimes contribute to saturation contrast, especially at a high pulse power setting, z-spectra of 80 μM protein samples using 279 μT saturation pulses were acquired and confirmed that the observed saturation contrast at this high pulse power is indeed due to interaction of the two fragments (Fig. S7, ESI†). Thus, regardless of fusion protein interactions, split TEM-1 has the potential to give background signal. This background was recognized in previous studies but thought to be negligible considering the high signal-to-noise ratio.2,3 Our results provide a cautionary note, showing that under conditions with high protein concentration (80 μM), the background signal resulting from spontaneous complementation makes split TEM-1 unsuitable for monitoring PPIs. This opens possibilities for engineering the split protein complementation reporter for studying weak PPIs.8
The hyper-CEST saturation contrast was then assessed in a cellular environment. E. coli strain BL21(DE3) cells co-expressing α and ω fragments were cultured in LB media, induced with isopropyl β-D-1-thiogalactopyranoside (IPTG), resuspended in PBS to an OD600 of 1, and tested by time-dependent saturation transfer experiments (Table 1 and Fig. S8, ESI†). Serving as negative controls, cells co-expressing the T1Fα/cJun-T1Fω pair with and without IPTG induction both reported saturation contrast of 0.12 ± 0.02 at +56 ppm. Similar background contrast was observed previously to originate from the nonspecific interaction of Xe with intracellular biomacromolecules.45,49–52E. coli cells co-expressing the interacting pair T1Fα-cFos/cJun-T1Fω showed saturation contrast of 0.18 ± 0.02 and 0.10 ± 0.02, with and without IPTG induction, respectively. The cFos/cJun interaction in E. coli cells reported saturation contrast of 0.08 after subtracting the non-induced background signal, and cFos deletion samples showed no change in saturation contrast, highlighting that 129Xe NMR can readily detect PPIs in bacterial cells.
| E. coli sample | T 1,on/s | T 1,off/s | Saturation contrast |
|---|---|---|---|
| T1Fα-cFos/cJun-T1Fω, non-induced | 16.0 ± 0.6 | 19.0 ± 0.7 | 0.10 ± 0.02 |
| T1Fα-cFos/cJun-T1Fω, induced | 16.0 ± 0.6 | 21.7 ± 0.7 | 0.18 ± 0.02 |
| T1Fα/cJun-T1Fω, non-induced | 14.1 ± 0.6 | 16.8 ± 0.3 | 0.12 ± 0.02 |
| T1Fα/cJun-T1Fω, induced | 17.1 ± 0.5 | 21.3 ± 0.6 | 0.12 ± 0.02 |
TEM-1 β-lactamase is non-toxic to prokaryotic and eukaryotic cells, and no orthologs of TEM-1 exist in eukaryotes. Therefore, the TEM-1-based complementation assay could be universally used in eukaryotic cells and many prokaryotes without intrinsic background activity. The ability of reconstituted (split) TEM-1 to bind xenon and report PPIs in vitro and in vivo using sensitive and selective hyper-CEST 129Xe NMR spectroscopy can expand the range of applications of PCAs in various biological systems. However, modest affinity between T1Fα and T1Fω fragments can result in background signal at high micromolar protein concentrations. It should be possible to reduce the intrinsic T1Fα/T1Fω interaction by disrupting several hydrogen bonds and salt bridges between T1Fα and T1Fω, which also has the potential to modulate Xe affinity. In enzymatic activity assays, the signal-to-noise ratio was improved by up to 104-fold via optimization of side chain contacts with the active site of TEM-1.3 Likewise, we envision that mutating Xe binding site residues can give enhancement to saturation contrast, as was previously demonstrated for maltose binding protein.49 Significant signal enhancement can also be achieved by use of isotopically enriched 129Xe and/or higher 129Xe hyperpolarization levels.53 Finally, the TEM-1 fragment complementation assay also holds promise for molecular recognition and selective turn-on sensing using 129Xe NMR/MRI, which has been a challenging task.49,54–57
We thank the University of Pennsylvania Chemistry NMR facility for spectrometer time. This work was supported by NIH grant R35-GM-131907.
Conflicts of interest
There are no conflicts to declare.Notes and references
- T. K. Kerppola, Nat. Methods, 2006, 3, 969–971 CrossRef CAS PubMed.
- A. Galarneau, M. Primeau, L. E. Trudeau and S. W. Michnick, Nat. Biotechnol., 2002, 20, 619–622 CrossRef CAS PubMed.
- T. Wehrman, B. Kleaveland, J. H. Her, R. F. Balint and H. M. Blau, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 3469–3474 CrossRef CAS PubMed.
- I. Ghosh, A. D. Hamilton and L. Regan, J. Am. Chem. Soc., 2000, 122, 5658–5659 CrossRef CAS.
- R. Paulmurugan and S. S. Gambhir, Anal. Chem., 2005, 77, 1295–1302 CrossRef CAS PubMed.
- R. Paulmurugan, Y. Umezawa and S. S. Gambhir, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 15608–15613 CrossRef CAS PubMed.
- J. N. Pelletier, F. X. Campbell-Valois and S. W. Michnick, Proc. Natl. Acad. Sci. U. S. A., 1998, 95, 12141–12146 CrossRef CAS PubMed.
- A. S. Dixon, M. K. Schwinn, M. P. Hall, K. Zimmerman, P. Otto, T. H. Lubben, B. L. Butler, B. F. Binkowski, T. Machleidt, T. A. Kirkland, M. G. Wood, C. T. Eggers, L. P. Encell and K. V. Wood, ACS Chem. Biol., 2016, 11, 400–408 CrossRef CAS PubMed.
- G. S. Loving, M. Sainlos and B. Imperiali, Trends Biotechnol., 2010, 28, 73–83 CrossRef CAS PubMed.
- T. K. Kerppola, Chem. Soc. Rev., 2009, 38, 2876–2886 RSC.
- X. Dai, M. Zhu and Y. P. Wang, Chem. Commun., 2014, 50, 1830–1832 RSC.
- T. Kakizuka, A. Takai, K. Yoshizawa, Y. Okada and T. M. Watanabe, Chem. Commun., 2020, 56, 3625–3628 RSC.
- I. Remy, G. Ghaddar and S. W. Michnick, Nat. Protoc., 2007, 2, 2302–2306 CrossRef CAS PubMed.
- E. Tchekanda, D. Sivanesan and S. W. Michnick, Nat. Methods, 2014, 11, 641–644 CrossRef CAS PubMed.
- B. R. Sculimbrene and B. Imperiali, J. Am. Chem. Soc., 2006, 128, 7346–7352 CrossRef CAS PubMed.
- E. Stefan, S. Aquin, N. Berger, C. R. Landry, B. Nyfeler, M. Bouvier and S. W. Michnick, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 16916–16921 CrossRef CAS PubMed.
- G. Loving and B. Imperiali, J. Am. Chem. Soc., 2008, 130, 13630–13638 CrossRef CAS PubMed.
- I. A. Demarco, A. Periasamy, C. F. Booker and R. N. Day, Nat. Methods, 2006, 3, 519–524 CrossRef CAS PubMed.
- I. Remy and S. W. Michnick, Nat. Methods, 2006, 3, 977–979 CrossRef CAS PubMed.
- C. D. Hu and T. K. Kerppola, Nat. Biotechnol., 2003, 21, 539–545 CrossRef CAS PubMed.
- S. A. Slavoff, D. S. Liu, J. D. Cohen and A. Y. Ting, J. Am. Chem. Soc., 2011, 133, 19769–19776 CrossRef CAS PubMed.
- E. Barile and M. Pellecchia, Chem. Rev., 2014, 114, 4749–4763 CrossRef CAS PubMed.
- E. R. Zuiderweg, Biochemistry, 2002, 41, 1–7 CrossRef CAS PubMed.
- L. D'Silva, P. Ozdowy, M. Krajewski, U. Rothweiler, M. Singh and T. A. Holak, J. Am. Chem. Soc., 2005, 127, 13220–13226 CrossRef PubMed.
- A. M. Bonvin, R. Boelens and R. Kaptein, Curr. Opin. Chem. Biol., 2005, 9, 501–508 CrossRef CAS PubMed.
- D. S. Burz, K. Dutta, D. Cowburn and A. Shekhtman, Nat. Methods, 2006, 3, 91–93 CrossRef CAS PubMed.
- M. P. Williamson, Prog. Nucl. Magn. Reson. Spectrosc., 2013, 73, 1–16 CrossRef CAS PubMed.
- M. L. Ludwiczek, B. Baminger and R. Konrat, J. Am. Chem. Soc., 2004, 126, 1636–1637 CrossRef CAS PubMed.
- M. Somlyay, K. Ledolter, M. Kitzler, G. Sandford, S. L. Cobb and R. Konrat, ChemBioChem, 2020, 21, 696–701 CrossRef CAS PubMed.
- P. Raffeiner, A. Schraffl, T. Schwarz, R. Rock, K. Ledolter, M. Hartl, R. Konrat, E. Stefan and K. Bister, Oncotarget, 2017, 8, 3327–3343 CrossRef PubMed.
- L. Schröder, T. J. Lowery, C. Hilty, D. E. Wemmer and A. Pines, Science, 2006, 314, 446–449 CrossRef PubMed.
- Y. Bai, P. A. Hill and I. J. Dmochowski, Anal. Chem., 2012, 84, 9935–9941 CrossRef CAS PubMed.
- Y. Wang and I. J. Dmochowski, Chem. Commun., 2015, 51, 8982–8985 RSC.
- M. G. Shapiro, R. M. Ramirez, L. J. Sperling, G. Sun, J. Sun, A. Pines, D. V. Schaffer and V. S. Bajaj, Nat. Chem., 2014, 6, 629–634 CrossRef CAS PubMed.
- T. K. Stevens, K. K. Palaniappan, R. M. Ramirez, M. B. Francis, D. E. Wemmer and A. Pines, Magn. Reson. Med., 2013, 69, 1245–1252 CrossRef CAS PubMed.
- T. K. Stevens, R. M. Ramirez and A. Pines, J. Am. Chem. Soc., 2013, 135, 9576–9579 CrossRef CAS PubMed.
- T. Meldrum, K. L. Seim, V. S. Bajaj, K. K. Palaniappan, W. Wu, M. B. Francis, D. E. Wemmer and A. Pines, J. Am. Chem. Soc., 2010, 132, 5936–5937 CrossRef CAS PubMed.
- L. Schröder, T. Meldrum, M. Smith, T. J. Lowery, D. E. Wemmer and A. Pines, Phys. Rev. Lett., 2008, 100, 257603 CrossRef PubMed.
- H. M. Rose, C. Witte, F. Rossella, S. Klippel, C. Freund and L. Schröder, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 11697–11702 CrossRef CAS PubMed.
- M. Kunth, J. Dopfert, C. Witte, F. Rossella and L. Schröder, Angew. Chem., Int. Ed., 2012, 51, 8217–8220 CrossRef CAS PubMed.
- M. Schnurr, K. Sydow, H. M. Rose, M. Dathe and L. Schröder, Adv. Healthcare Mater., 2015, 4, 40–45 CrossRef CAS PubMed.
- L. J. Ransone, J. Visvader, P. Sassonecorsi and I. M. Verma, Genes Dev., 1989, 3, 770–781 CrossRef CAS PubMed.
- J. C. Saunders, L. M. Young, R. A. Mahood, M. P. Jackson, C. H. Revill, R. J. Foster, D. A. Smith, A. E. Ashcroft, D. J. Brockwell and S. E. Radford, Nat. Chem. Biol., 2016, 12, 94–101 CrossRef CAS PubMed.
- C. K. Zhao, Q. Yin and S. Y. Li, Acta Pharmacol. Sin., 2010, 31, 1618–1624 CrossRef CAS PubMed.
- Y. Wang, B. W. Roose, E. J. Palovcak, V. Carnevale and I. J. Dmochowski, Angew. Chem., Int. Ed., 2016, 55, 8984–8987 CrossRef CAS PubMed.
- B. W. Roose, S. D. Zemerov, Y. Wang, M. A. Kasimova, V. Carnevale and I. J. Dmochowski, ChemPhysChem, 2019, 20, 260–267 CrossRef CAS PubMed.
- T. D. Halazonetis, K. Georgopoulos, M. E. Greenberg and P. Leder, Cell, 1988, 55, 917–924 CrossRef CAS PubMed.
- N. Szaloki, J. W. Krieger, I. Komaromi, K. Toth and G. Vamosi, Mol. Cell. Biol., 2015, 35, 3785–3798 CrossRef CAS PubMed.
- B. W. Roose, S. D. Zemerov and I. J. Dmochowski, Chem. Sci., 2017, 8, 7631–7636 RSC.
- C. Boutin, H. Desvaux, M. Carriere, F. Leteurtre, N. Jamin, Y. Boulard and P. Berthault, NMR Biomed., 2011, 24, 1264–1269 CrossRef CAS PubMed.
- K. K. Palaniappan, R. M. Ramirez, V. S. Bajaj, D. E. Wemmer, A. Pines and M. B. Francis, Angew. Chem., Int. Ed., 2013, 52, 4849–4853 CrossRef CAS PubMed.
- A. Bifone, Y. Q. Song, R. Seydoux, R. E. Taylor, B. M. Goodson, T. Pietrass, T. F. Budinger, G. Navon and A. Pines, Proc. Natl. Acad. Sci. U. S. A., 1996, 93, 12932–12936 CrossRef CAS PubMed.
- P. Nikolaou, A. M. Coffey, L. L. Walkup, B. M. Gust, N. Whiting, H. Newton, S. Barcus, I. Muradyan, M. Dabaghyan, G. D. Moroz, M. S. Rosen, S. Patz, M. J. Barlow, E. Y. Chekmenev and B. M. Goodson, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 14150–14155 CrossRef CAS PubMed.
- C. C. Slack, J. A. Finbloom, K. Jeong, C. J. Bruns, D. E. Wemmer, A. Pines and M. B. Francis, Chem. Commun., 2017, 53, 1076–1079 RSC.
- J. A. Finbloom, C. C. Slack, C. J. Bruns, K. Jeong, D. E. Wemmer, A. Pines and M. B. Francis, Chem. Commun., 2016, 52, 3119–3122 RSC.
- N. Kotera, N. Tassali, E. Leonce, C. Boutin, P. Berthault, T. Brotin, J. P. Dutasta, L. Delacour, T. Traore, D. A. Buisson, F. Taran, S. Coudert and B. Rousseau, Angew. Chem., Int. Ed., 2012, 51, 4100–4103 CrossRef CAS PubMed.
- N. Tassali, N. Kotera, C. Boutin, E. Leonce, Y. Boulard, B. Rousseau, E. Dubost, F. Taran, T. Brotin, J. P. Dutasta and P. Berthault, Anal. Chem., 2014, 86, 1783–1788 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures, sample characterizations and hyper-CEST data. See DOI: 10.1039/d0cc02988b |
| This journal is © The Royal Society of Chemistry 2020 |