
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthesis of protected 3-aminopiperidine and 3-aminoazepane derivatives using enzyme cascades†
Grayson J.
Ford
,
Nico
Kress
,
Ashley P.
Mattey
 ,
Lorna J.
Hepworth
,
Christopher R.
Baldwin
,
James R.
Marshall
,
Lisa S.
Seibt
,
Min
Huang
,
William R.
Birmingham
,
Nicholas J.
Turner
and
Sabine L.
Flitsch
*
,
Lorna J.
Hepworth
,
Christopher R.
Baldwin
,
James R.
Marshall
,
Lisa S.
Seibt
,
Min
Huang
,
William R.
Birmingham
,
Nicholas J.
Turner
and
Sabine L.
Flitsch
*
Department of Chemistry, Manchester Institute of Biotechnology, University of Manchester, 131 Princess Street, Manchester, M1 7DN, UK. E-mail: Sabine.flitsch@manchester.ac.uk
First published on 2nd June 2020
Abstract
Multi-enzyme cascades utilising variants of galactose oxidase and imine reductase led to the successful conversion of N-Cbz-protected L-ornithinol and L-lysinol to L-3-N-Cbz-aminopiperidine and L-3-N-Cbz-aminoazepane respectively, in up to 54% isolated yield. Streamlining the reactions into one-pot prevented potential racemisation of key labile intermediates and led to products with high enantiopurity.
Chiral amine moieties are present in many of the most valuable pharmaceutical compounds,1,2 with cyclic diamines in particular often used as semi-rigid bifunctional linkers in medicinal chemistry. Hence there is a demand for efficient, stereoselective synthesis strategies of diamines from easily accessible starting materials, preferentially in their semi-protected form to allow direct application in the manufacture of pharmaceutical intermediates. Recent biocatalytic methodologies have proven to be very successful in the design of alternative efficient and sustainable processes in the synthesis of chiral amines, underpinned by an increasing interest in their industrial application.3 In this regard, we were interested in the chiral enzymatic synthesis of semi-protected 3-aminopiperidines 7 and 3-aminoazepanes 8, being core structures in many valuable pharmaceutical drugs such as alogliptin and besifloxacin.4,5
Current synthetic methods towards 3-aminopiperidines and 3-aminoazepanes encompass various approaches, including Curtius and Hofmann rearrangements, hydrogenation of 3-aminopyridine or the cyclisation of α-amino acids.6–8 Although these routes are well established, they often lack chiral control and the use of expensive and toxic reagents can hinder their wider industrial application. The particular challenge of chiral 3-aminoazepanes such as 8 was recently addressed by Feng et al. using a transaminase-based biocatalytic synthesis of 3-aminoazepane from ketone precursors.9 An attractive strategy is the combination of biocatalysts in telescoped enzymatic cascades resembling biosynthetic pathways to generate unnatural compounds.10–13
When targeting 7 and 8, the challenge was to find enzymes that could tolerate bulky unnatural protection groups such as carboxybenzyl (Cbz) groups, which are needed for subsequent coupling chemistries. Based on our previous expertise in the application of galactose oxidase (GOase)14–17 and imine reductase (IRED)18–22 biocatalysts, a multi-enzymatic cascade towards amino-piperidine and amino azepanes was designed. This streamlined approach uses readily available amino alcohols such as 1a–c and 2a–c as substrates, (Fig. 1), which were directly accessible by chemical synthesis from natural amino acids L-ornithine and L-lysine (ESI†).
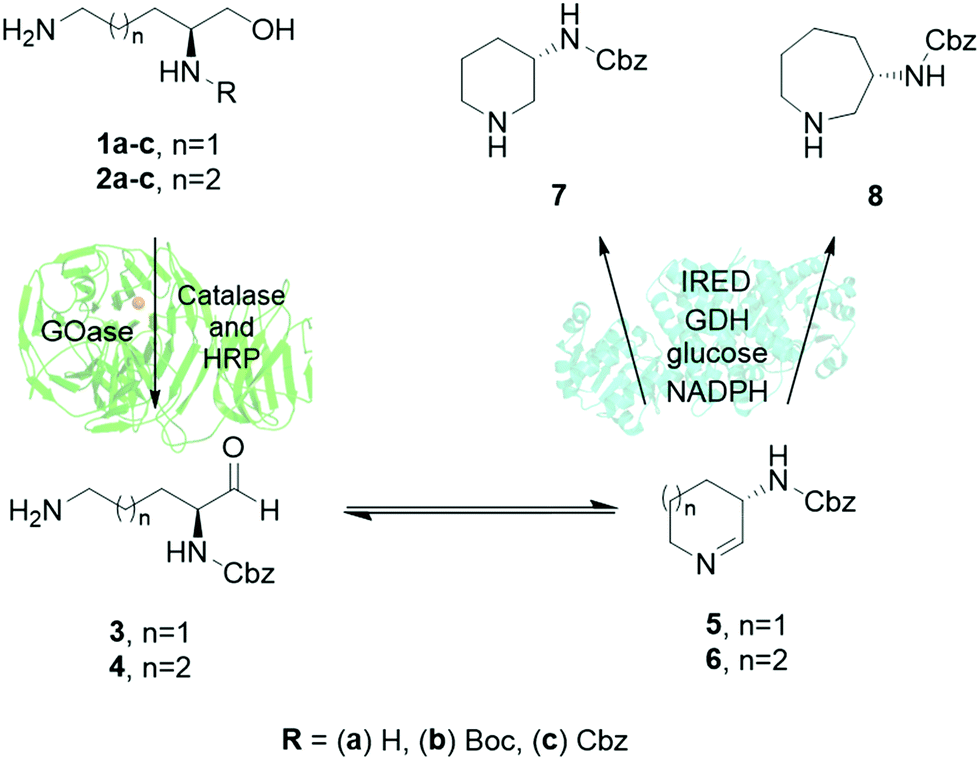 | ||
| Fig. 1 Enzyme cascade for the synthesis of L-3-N-aminopiperidines and L-3-N-aminoazepanes using a combination of galactose oxidase (GOase) and imine reductase (IRED) enzymes. | ||
The enzymatic cascade would then proceed by initial oxidation of amino alcohols 1 and 2 to the respective amino aldehydes 3 or 4 by GOase, followed by spontaneous formation of cyclic imine intermediates 5 or 6 that would finally be reduced by an IRED to generate the desired enantiopure products 7 and 8.17 Both individual enzymatic reactions have already shown promise in industrial applications.23,24
The proposed strategy presented multiple challenges: the α-amino aldehyde intermediates from substrates 1a and 2a would be predicted to be very unstable in a biotransformation. Considering the need for selective protection of the target compounds for subsequent chemical coupling reactions in API synthesis anyway, Boc- and Cbz-protected starting materials 1b–c and 2b–c were investigated as leading to reagents that are more valuable. However, the use of monoprotected substrates would require the tolerance of the chosen biocatalysts towards bulky hydrophobic protecting groups. To minimize side-reactions, purified enzymes rather than whole cells or enzyme lysates were used.
For the first step of the enzyme cascade, suitable recombinant GOase variants F2 and M3–5 were tested for the oxidation of amino alcohols 1a–c and 2a–c (ESI†).14,17 An initial activity screen showed that the Cbz-protected amino alcohol derivatives 1c and 2c gave highest initial activity, whilst unprotected and Boc-protected derivatives 1a–b and 2a–b were not well accepted as substrates (ESI†). The poor results for the unprotected substrates 1a and 2a might be due to instability of the amino aldehyde intermediates and problems with amino alcohol functionality, which can result in potential chelation of the active copper center in GOase.25
Given that Cbz-protected substrates 1c and 2c demonstrated good activity, a colorimetric HRP-ABTS assay was used to determine kinetic constants comparing the two GOase mutants F2 and M3–5 (Table 1).26 Both mutants showed overall comparable activity, L-lysinol derivative 2c appeared to be preferred over L-ornithinol 1c, with significant higher catalytic efficiency, particularly for the M3–5 variant.
| Substrate–GOase | K m [mM] | V max [U mg−1] | k cat [s−1] | K cat/Km [mM s−1] |
|---|---|---|---|---|
| Conditions: ABTS-HRP coupled assay in NaPi (100 mM, pH 7.4) with substrates 1c and 2c at various concentrations, λ = 420 nm, 25 °C (ESI). | ||||
| 1c–F2 | 70.7 ± 7.2 | 0.83 ± 0.09 | 0.9 ± 0.1 | 0.013 |
| 1c–M3–5 | 47 ± 15 | 0.77 ± 0.22 | 0.87 ± 0.25 | 0.019 |
| 2c–F2 | 70.5 ± 2.2 | 1.74 ± 0.05 | 1.97 ± 0.06 | 0.028 |
| 2c–M3–5 | 40.5 ± 7.3 | 2.29 ± 0.34 | 2.57 ± 0.42 | 0.063 |
Encouraged by these results, the GOase variants were tested in combination with an IRED panel in a one-pot reaction, thus avoiding the need to isolate potentially unstable aldehyde/imine intermediates. Five IREDs were selected from available libraries for the first screen: AdRedAm, IR-23, IR-49, IR-102 and IR-110. AdRedAm was chosen because of its broad substrate range, whereas the other four IREDs have been previously shown to accept the azepane scaffold.20,27
Previous studies directed the reaction design for the one-pot in vitro GOase–IRED cascade, which included HRP for activation of GOase, catalase for H2O2 decomposition, while NADP+ in combination with glucose dehydrogenase (GDH) and glucose was added as a cofactor recycling system to ensure electron supply for the IRED reaction.26,28 Considering previously optimized reaction conditions for both GOase and IRED, initial analytical scale reactions were performed in phosphate buffer at pH 7.5 and 30 °C for 16 h.26,29
Chromatographic analysis of the reaction mixtures showed only trace amounts of products for 1a and 2a, and so were not investigated further (ESI†). The Boc-protected substrates 1b and 2b showed no detectable activity, but we were pleased to observe a range of potential GOase–IRED combinations to be active towards the Cbz-protected substrates 1c and 2c (Fig. 2 and ESI†). We were surprised to find a marked preference for the L-lysinol substrate 2c over the L-ornithinol substrate 1c, considering that the azepane imine intermediate would be expected to be less readily formed than the corresponding piperidine imine. For both substrates, the GOase M3–5 variant performed considerably better than F2, which is in agreement with previously reported amino alcohol conversions.17 The IRED preference was found to be strongly substrate dependent, with AdRedAm performing best in the case of the piperidine product 7, while IR-49 was optimal for azepane 8 synthesis.
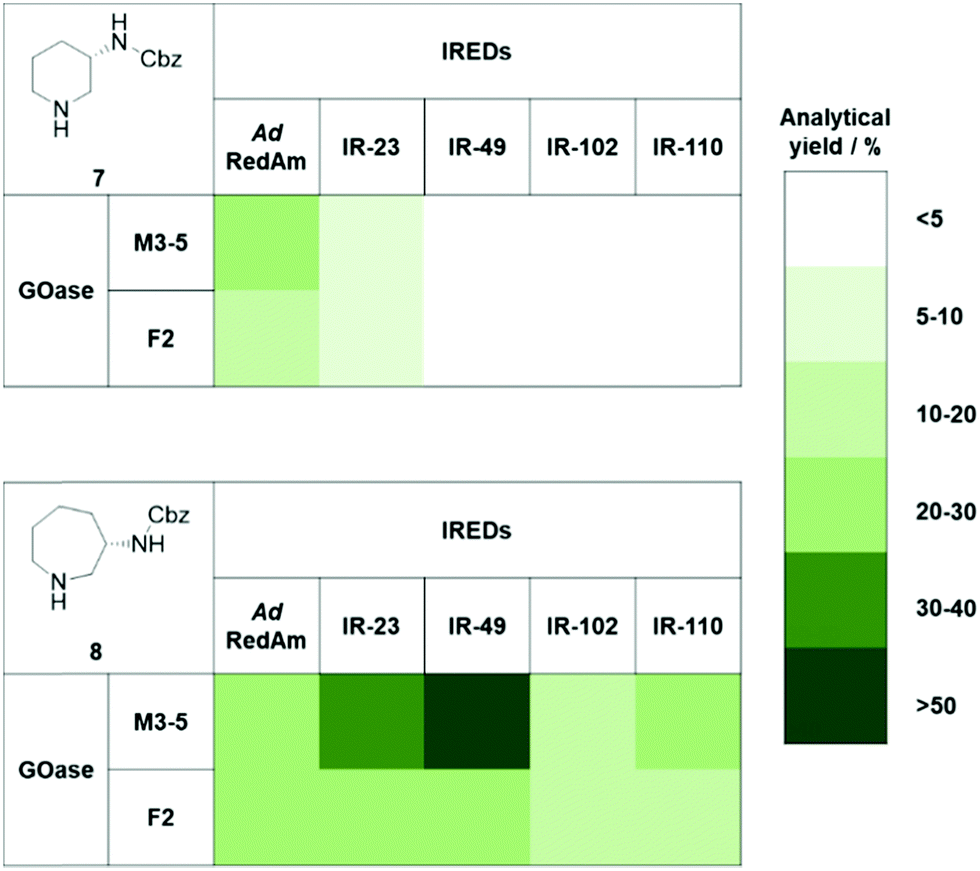 | ||
| Fig. 2 Result of activity screening for the synthesis of 3-N-Cbz-aminopiperidine 7 and 3-N-Cbz-aminoazepane 8 comparing 2 GOases and 5 IREDs. Reactions were performed with 3 mM substrate in NaPi buffer (pH 7.5) at 30 °C and 200 rpm for 16 h. The resulting analytical yields were determined by GC-FID and are given as colour-coded ranges. | ||
A set of reaction optimizations were performed for both cascades to give products 7 and 8, including pH, reaction temperature, enzyme loading and substrate concentration (ESI†). Each parameter was altered individually while other parameters stayed constant for both. The cascade was screened over a range of reaction pHs (6–8). An optimal pH of 8 was determined for the enzyme cascades, resulting in higher product yields for both when compared to a lower reaction pH, following general previous trends for both enzymes used.26,29 Increasing reaction temperatures above 30 °C resulted in lower conversions, suggesting deactivation of the enzyme at these temperatures. Increasing IRED concentration showed minimal variation in the yield, but increasing the GOase concentration improved product yield, confirming that the IRED does not appear to be rate limiting. It was also noted that conversions for the transformation did not increase further when more than 0.5 mg ml−1 GOase was used for the transformation of 2c to product 8, but did increase up to 1 mg ml−1 for the transformation of 1c to product 7. Combined with the initial kinetic screening results, this further confirms the higher affinity GOase M3–5 has for substrate 2c over 1c.
Given that the GOase is the gatekeeper for the cascade, molecular docking studies were performed to better understand substrate specificity. F2 and M3–5 models were created based on the reported E1 variant structure (PDB 2WQ8). 1c and 2c were used as ligands for the docking simulation into the two GOase mutants. The displayed binding modes in Fig. 3 represent the catalytic binding modes with the highest calculated binding affinity. For catalytic binding, the free copper binding site needs to be occupied by the hydroxy group that is to be oxidized in the radical-based GOase mechanism.30 In the E1 crystal structure, a co-crystallized acetate binds the copper in 2.3 Å distance, which is closely mimicked by the calculated binding of 1c and 2c to M3–5. In contrast, more than 1 Å greater distances are calculated for binding to F2, supporting the kinetic data. Interestingly, the ligands adopt two opposing binding modes with respect to the Cbz phenyl group.
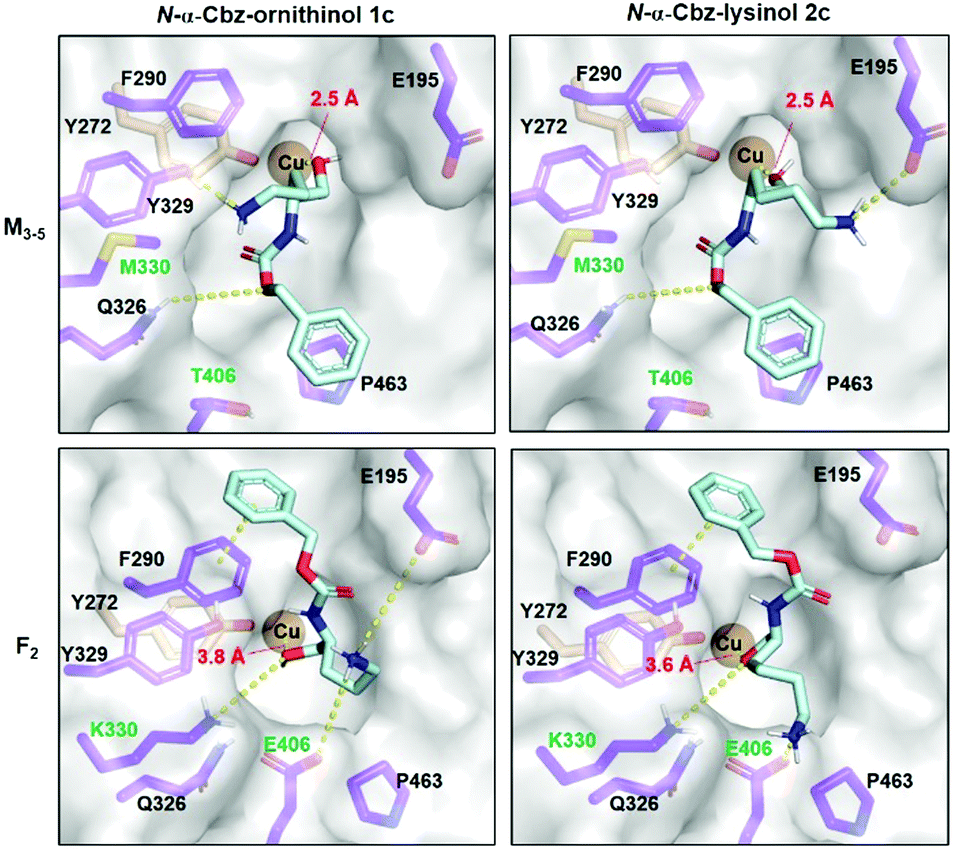 | ||
| Fig. 3 Molecular docking (AutoDock VINA as implemented in YASARA) of N-α-Cbz-ornithinol 1c and N-α-Cbz-lysinol 2c in GOase variants M3–5 and F2 modelled based on the E1 variant crystal structure (PDB 2WQ8). Substrate (cyan) conformations positioning the target hydroxyl group towards the copper centre (dark orange) are depicted with the respective O–Cu distance in red. Substrate-interacting active site amino acids are highlighted (purple) indicating residues altered in between the GOase variants (green). Selected substrate–receptor interactions are highlighted (yellow). | ||
While the phenyl group binds atop P463 in M3–5, it is bound above F290 in F2 allowing for stronger π–π stacking. Two of the mutational differences between M3–5 and F2 seem to be mainly responsible for the altered interaction networks in substrate binding being M/K330 and T/E406. K330 in F2 seems to cause a displaced hydroxy orientation, while E406 is a potential salt bridge partner for the terminal charged amino group. Comparing 1c and 2c, the extended length of the lysinol allows for a strong salt bridge formation with E195 and E406 in M3–5 and F2 respectively; which is less pronounced for the shorter ornithinol, serving as a potential structural explanation for the observed preference for lysinol.
Using optimized reaction conditions of higher GOase concentrations and a reaction pH of 8, the biotransformations of 1c and 2c were scaled up to a final volume of 90 mL. For substrate 1c (3 mM), GOase M3–5 and AdRedAm were used (1 mg ml−1 and 0.25 mg ml−1 respectively). Protein precipitation of AdRedAm was observed after 1–3 h of the reaction in contrast to the analytical scale reactions (ESI†). Nonetheless, we were able to extract and isolate product 7 in 16% yield (10 mg) using a simple acid–base wash. For substrate 2c, we were able to use less enzyme with 0.5 mg ml−1 M3–5 and 0.25 mg ml−1 IR-49. Under these reaction conditions, no precipitate formed within the 48 h of reaction time and following the same workup protocol as before, a yield of 54% (36 mg) product 8 was isolated and characterized from the reaction.
The enantiopurity of 7 and 8 was determined using chiral normal phase HPLC analysis, comparing the products from the scaled-up biotransformations, with authentic and synthesized standards (ESI†). Chromatograms showed the presence of only one enantiomer per reaction, confirming preservation of the initial amino acid-derived L-configuration.
In conclusion, we showed that industrially relevant enzymes, GOase and IRED can be combined in a novel one-pot enzymatic cascade to synthesize enantiopure Cbz-protected L-3-aminopiperidine 7 and L-3-aminoazepane 8 products from amino alcohols derived from bio-renewable amino acids as a green feedstock under ambient conditions. The strategy allows for directly generating selectively Cbz protected targets, that are compatible for subsequent incorporation into chemical processes. The lack of racemization demonstrates the advantage of using one-pot streamlined cascades.
We kindly acknowledge financial support from the European Research Council (788231-ProgrES-ERC-2017-ADG to SLF; BIO-H-BORROW: Grant No. 742987 to NJT); EU (CarboMet 737395) BBSRC (BB/M027791/1; BB/M02903411; BB/M028836/1). J. R. M. acknowledges the Industrial Biotechnology Innovation Centre (IBioIC) and BBSRC funding through a CASE studentship with Prozomix Ltd. W. R. B. gratefully acknowledges the Biotechnology and Biological Sciences Research Council (BBSRC) for the award of a Discovery Fellowship (BB/S010459/1).
Conflicts of interest
There are no conflicts to declare.Notes and references
- M. Breuer, K. Ditrich, T. Habicher, B. Hauer, M. Keßeler, R. Stürmer and T. Zelinski, Angew. Chem., Int. Ed., 2004, 43, 788–824 CrossRef CAS PubMed.
- D. Ghislieri and N. J. Turner, Top. Catal., 2014, 57, 284–300 CrossRef CAS.
- M. Schober, C. MacDermaid, A. A. Ollis, S. Chang, D. Khan, J. Hosford, J. Latham, L. A. F. Ihnken, M. J. B. Brown, D. Fuerst, M. J. Sanganee and G. D. Roiban, Nat. Catal., 2019, 2, 909–915 CrossRef CAS.
- J. Feng, Z. Zhang, M. B. Wallace, J. A. Stafford, S. W. Kaldor, D. B. Kassel, M. Navre, L. Shi, R. J. Skene, T. Asakawa, K. Takeuchi, R. Xu, D. R. Webb and S. L. Gwaltney, J. Med. Chem., 2007, 50, 2297–2300 CrossRef CAS PubMed.
- W. Haas, C. M. Pillar, G. E. Zurenko, J. C. Lee, L. S. Brunner and T. W. Morris, Antimicrob. Agents Chemother., 2009, 53, 3552–3560 CrossRef CAS PubMed.
- H. Nienburg, Ber. Dtsch. Chem. Ges. A/B, 1937, 70, 635–638 CrossRef.
- M. Yamada, S. Hirano, R. Tsuruoka and M. Yamano, WO 2008/102720 A1, 2008.
- K. Oskada, T. Ikariya, M. Saburi and S. Yoshikawa, Chem. Lett., 1981, 1691–1694 CrossRef.
- Y. Feng, Z. Luo, G. Sun, M. Chen, J. Lai, W. Lin, S. Goldmann, L. Zhang and Z. Wang, Org. Process Res. Dev., 2017, 21, 648–654 CrossRef CAS.
- S. P. France, L. J. Hepworth, N. J. Turner and S. L. Flitsch, ACS Catal., 2017, 7, 710–724 CrossRef CAS.
- J. H. Schrittwieser, S. Velikogne, M. Hall and W. Kroutil, Chem. Rev., 2018, 118, 270–348 CrossRef CAS PubMed.
- J. I. Ramsden, R. S. Heath, S. R. Derrington, S. L. Montgomery, J. Mangas-Sanchez, K. R. Mulholland and N. J. Turner, J. Am. Chem. Soc., 2019, 141, 1201–1206 CrossRef CAS PubMed.
- C. Martin, M. Trajkovic and M. W. Fraaije, Angew. Chem., Int. Ed., 2020, 59, 4869–4872 CrossRef CAS PubMed.
- L. Sun, I. P. Petrounia, M. Yagasaki, G. Bandara and F. H. Arnold, Protein Eng., Des. Sel., 2001, 14, 699–704 CrossRef CAS PubMed.
- J. B. Rannes, A. Ioannou, S. C. Willies, G. Grogan, C. Behrens, S. L. Flitsch and N. J. Turner, J. Am. Chem. Soc., 2011, 133, 8436–8439 CrossRef CAS PubMed.
- F. Escalettes and N. J. Turner, ChemBioChem, 2008, 9, 857–860 CrossRef CAS PubMed.
- A. J. Carnell, A. R. Frazer, S. M. McKenna, S. Herter, S. Leimkühler and N. J. Turner, ChemCatChem, 2015, 7, 2313–2317 CrossRef.
- K. Mitsukura, M. Suzuki, K. Tada, T. Yoshida and T. Nagasawa, Org. Biomol. Chem., 2010, 8, 4533–4535 RSC.
- M. Lenz, N. Borlinghaus, L. Weinmann and B. M. Nestl, World J. Microbiol. Biotechnol., 2017, 33, 199 CrossRef PubMed.
- W. Zawodny, S. L. Montgomery, J. R. Marshall, J. D. Finnigan, N. J. Turner and J. Clayden, J. Am. Chem. Soc., 2018, 140, 17872–17877 CrossRef CAS PubMed.
- P. Matzel, M. Gand and M. Höhne, Green Chem., 2017, 19, 385–389 RSC.
- A. Al-Shameri, N. Borlinghaus, L. Weinmann, P. N. Scheller, B. M. Nestl and L. Lauterbach, Green Chem., 2019, 21, 1396–1400 RSC.
- M. A. Huffman, A. Fryszkowska, O. Alvizo, M. Borra-Garske, K. R. Campos, K. A. Canada, P. N. Devine, D. Duan, J. H. Forstater, S. T. Grosser, H. M. Halsey, G. J. Hughes, J. Jo, L. A. Joyce, J. N. Kolev, J. Liang, K. M. Maloney, B. F. Mann, N. M. Marshall, M. McLaughlin, J. C. Moore, G. S. Murphy, C. C. Nawrat, J. Nazor, S. Novick, N. R. Patel, A. Rodriguez-Granillo, S. A. Robaire, E. Sherer, M. D. Truppo, A. M. Whittaker, D. Verma, L. Xiao, Y. Xu and H. Yang, Science, 2019, 366, 1255–1259 CrossRef CAS PubMed.
- M. Schober, C. MacDermaid, A. A. Ollis, S. Chang, D. Khan, J. Hosford, J. Latham, L. A. F. Ihnken, M. J. B. Brown, D. Fuerst, M. J. Sanganee and G. D. Roiban, Nat. Catal., 2019, 2, 909–915 CrossRef CAS.
- N. Kaewchangwat, S. Dueansawang, G. Tumcharern and K. Suttisintong, J. Agric. Food Chem., 2017, 65, 9828–9837 CrossRef CAS PubMed.
- A. Toftgaard Pedersen, W. R. Birmingham, G. Rehn, S. J. Charnock, N. J. Turner and J. M. Woodley, Org. Process Res. Dev., 2015, 19, 1580–1589 CrossRef CAS.
- G. A. Aleku, S. P. France, H. Man, J. Mangas-Sanchez, S. L. Montgomery, M. Sharma, F. Leipold, S. Hussain, G. Grogan and N. J. Turner, Nat. Chem., 2017, 9, 961–969 CrossRef CAS PubMed.
- C. H. Wong, D. G. Drueckhammer and H. M. Sweers, J. Am. Chem. Soc., 1985, 107, 4028–4031 CrossRef CAS.
- J. Mangas-Sanchez, S. P. France, S. L. Montgomery, G. A. Aleku, H. Man, M. Sharma, J. I. Ramsden, G. Grogan and N. J. Turner, Curr. Opin. Chem. Biol., 2017, 37, 19–25 CrossRef CAS PubMed.
- R. M. Wachter and B. P. Branchaud, J. Am. Chem. Soc., 1996, 118, 2782–2789 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0cc02976a |
| This journal is © The Royal Society of Chemistry 2020 |