
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A short peptide that preferentially binds c-MYC G-quadruplex DNA†
Aisling
Minard‡
 ab,
Danielle
Morgan‡
c,
Federica
Raguseo
a,
Anna
Di Porzio
ad,
Denise
Liano
ab,
Andrew G.
Jamieson
*c and
Marco
Di Antonio
*ab
ab,
Danielle
Morgan‡
c,
Federica
Raguseo
a,
Anna
Di Porzio
ad,
Denise
Liano
ab,
Andrew G.
Jamieson
*c and
Marco
Di Antonio
*ab
aImperial College London, Chemistry Department, Molecular Science Research Hub, 80 Wood Lane, W12 0BZ, London, UK. E-mail: m.di-antonio@imperial.ac.uk
bInstitute of Chemical Biology, Molecular Science Research Hub, 80 Wood Lane, W12 0BZ, London, UK
cSchool of Chemistry, University of Glasgow, G12 8QQ, Glasgow, UK. E-mail: andrew.jamieson.2@glasgow.ac.uk
dDepartment of Pharmacy, University of Naples Federico II, Via D. Montesano 49, 80131 Naples, Italy
First published on 1st July 2020
Abstract
G-quadruplexes (G4s) are non-canonical DNA secondary structures. The identification of selective tools to probe individual G4s over the ∼700![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 found in the human genome is key to unravel the biological significance of specific G4s. We took inspiration from a crystal structure of the bovine DHX36 helicase bound to the G4 formed in the promoter region of the oncogene c-MYC to identify a short peptide that preferentially binds MYC G4 with nM affinity over a small panel of parallel and non-parallel G4s tested.
000 found in the human genome is key to unravel the biological significance of specific G4s. We took inspiration from a crystal structure of the bovine DHX36 helicase bound to the G4 formed in the promoter region of the oncogene c-MYC to identify a short peptide that preferentially binds MYC G4 with nM affinity over a small panel of parallel and non-parallel G4s tested.
Besides the well-known double-helical structure, DNA can adopt several non-canonical structural arrangements under physiological conditions.1 G-quadruplexes (G4s) have recently emerged as an interesting alternative DNA structure with respect to their potential for biological regulation. Sequences that form G4s are highly abundant, with more than 700
000 G4-structures experimentally detected across the human genome by sequencing experiments.2 Because of their high genomic prevalence, DNA G4s have been speculated to play a role in several biological processes, including transcriptional regulation,3 telomeric maintenance,4 genomic instability,5 cancer progression,3 accelerating ageing6 and neurodegenerative diseases.7 To this end, several tools to map, visualise and stabilise G4s in biologically relevant contexts have been developed, in an attempt to unravel the specific biological roles played by G4-formation in cells.8 Although genome-wide mapping experiments by means of Chromatin Immuno-Precipitation (ChIP) methods have facilitated the detection of G4s that are actually formed in cells,9 the investigation of biological processes regulated by G4-stabilisation mainly relies on the use of small-molecule ligands that can be applied in living cells and monitored in real-time. To date, several ligands with high affinity and specificity for G4s over canonical double-stranded DNA have been reported.10 Small-molecule targeting approaches mainly rely on π–π stacking interactions of the molecular probes with the top (or bottom) end of the G4-structure. Whilst this approach is highly effective to achieve G4 vs. duplex selectivity, it is less applicable to achieve selectivity towards a small subset of G4s over the ∼700000 available in the human genome. This is most likely due to the promiscuous presence of accessible π-stacking surfaces across different G4-structures, which hampers selective recognition of an individual G4 by means of π–π stacking interactions alone.
Here, we have taken inspiration from a recently reported crystal structure of the bovine DHX36 helicase bound to the G4-structure formed in the promoter region of the oncogene c-MYC11 to identify a short-peptide sequence (DM039) that binds with nM affinity the MYC G4. Interestingly, DM039 showed negligible binding to other G4-forming sequences of parallel, antiparallel and mixed type topologies, suggesting potential for selective targeting of MYC. Furthermore, we found that when constrained into a helical conformation by hydrocarbon stapling, the peptide displayed lower binding affinity to MYC and loss of selectivity towards double- and single-stranded DNA. This data suggested that the peptide requires a good degree of structural flexibility for high affinity and selective G4-binding, which is not intuitive based on the helical conformation adopted by the peptide in the crystal structure.11
We started our investigation analysing the crystal structure of the bovine DHX36 bound to MYC G411 (PDB ID: 5VHE) using PyMOL12 (Fig. 1A) and identified the 22-amino acid sequence reported in Fig. 1B as the minimal MYC-binding domain of the protein in this particular context. This 22-amino acid sequence (Fig. 1B) is in agreement with that previously reported by NMR studies on the human version of the DHX36 helicase, which identified similar sequences (18, 23, 29 and 53 amino acids) as G4-binding motifs.13 We then synthesised the 22-amino acids sequence (DM039) to investigate whether this particular peptide motif displayed an increased selectivity towards MYC G4 over other G4-structures. DM039 was synthesised using previously established Fmoc/tBu solid-phase peptide synthesis (SPPS) protocols, employing an automated SPPS synthesiser, and acetylated at the N-terminal prior to HPLC purification and MS characterisation (see ESI†).
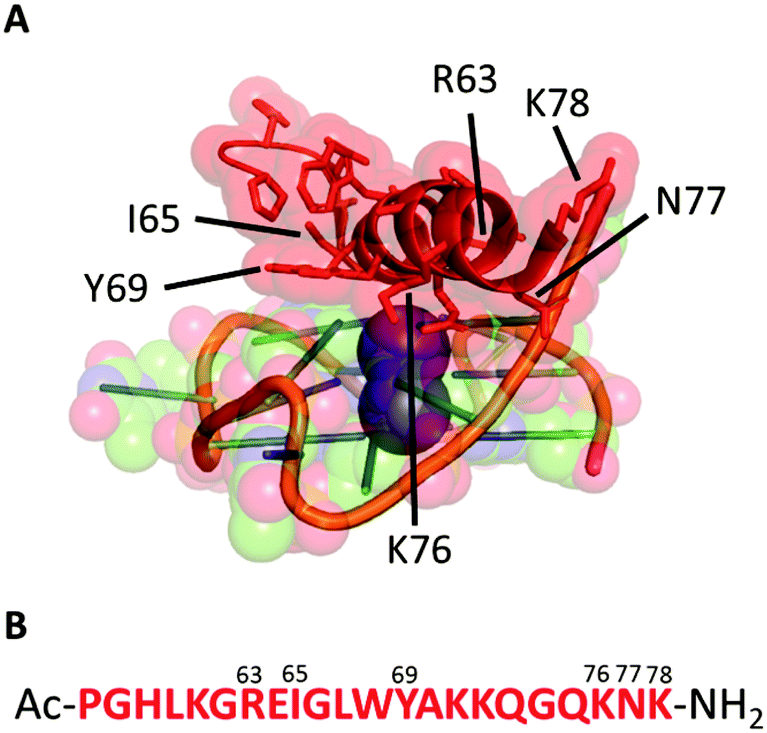 | ||
| Fig. 1 (A) Minimal peptide binding domain of DHX36 bound to the MYC DNA G-quadruplex. (B) The 22-amino acid sequence (DM039) used in this study. | ||
We next sought to evaluate the binding affinity of DM039 towards a small panel of G4-forming sequences as well as a single-stranded and a double-stranded control sequence. Typically, small-molecule ligands that bind G4s are tested by melting experiments (FRET-melting) for their ability to stabilise G4-structures.14 Even though stabilisation properties are correlated, on some level, to the binding affinity of the ligands, they cannot be considered a direct affinity measurement and are difficult to apply to peptides that are temperature sensitive. To overcome this limitation, we decided to exploit the higher molecular weight of the 22-amino acid peptide, compared to small-molecules, to develop a Fluorescence-Polarisation (FP) assay to reliably measure G4-binding. The FP measurements rely on the use of a fluorescein (FAM)-labelled oligonucleotide and the formation of a big complex (peptide–DNA) upon binding. The peptide–DNA complex formation causes a slower tumbling of the FAM-labelled oligonucleotide, which in turn generates a fluorescence polarised signal that can be detected as a function of the fraction of the oligonucleotide bound to the peptide.15 We initially applied our FP-assay to evaluate the binding affinity of DM039 to MYC G4. As displayed in Fig. 2, DM039 binds with high affinity to MYC G4 with an observed Kd of 112 nM [59–196 nM – 95% CI], confirming the suitability of our FP-assay to measure the binding affinity of short peptides to G4s. We next tested whether the high binding affinity of DM039 to MYC was specific to G4-folded oligonucleotides, testing its binding by FP to a FAM-labelled single-stranded mutant of MYC, no longer able to fold into a G4, and to a FAM-labelled double-stranded DNA sequence (see ESI†). DM039 displayed negligible binding to both the MYC-mutant and double-stranded DNA controls, suggesting that G4-specific interactions are responsible for the observed binding to the MYC G4. To further test that the binding to MYC was dependent on the G4-folding status, we measured reduced binding affinity of DM039 to the MYC G4 sequence when annealed in Li+ buffer, which prevents stabilisation of the formed G4. The resulting Kd was 1226 nM [491–3469 nM – 95% CI], proving a ∼10-fold reduction in binding affinity with respect to the binding measured in K+ buffer (see ESI†).
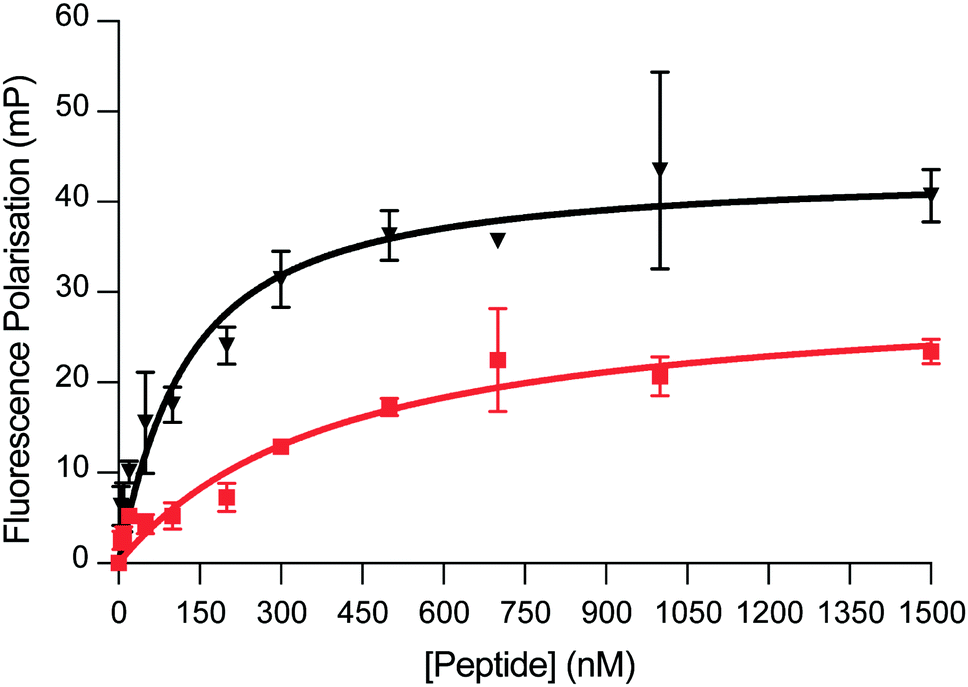 | ||
| Fig. 2 FP binding traces obtained with DM039 in the presence of c-MYC (black) and BCL-2 (red). The FP experiments were performed in Inner cell buffer (see ESI†), at room temperature, by using a fixed concentration of oligomer (20 nM) and increasing concentrations of peptide (0–5000 nM. Here, only the range 0–1500 nM is shown) (see ESI†). Each measurement was repeated three times. | ||
Since previous reports on similar peptide sequences suggested selectivity for parallel G4s over antiparallel ones,13 we decided to test the binding affinity of DM039 to other parallel G4s. Specifically, we have tested FP-binding of DM039 to FAM-labelled c-KIT1 and c-KIT2 G4s, which are known to fold into a parallel topology.16 Surprisingly, DM039 showed negligible binding to both c-KIT1 and c-KIT2 G4s (see ESI†), suggesting that this particular peptide sequence is not selective to all parallel conformations but can strongly discriminate against G4s of the same topology.
We further investigated whether DM039 was unable to bind non-parallel G4s, as reported for similar peptide sequences extracted from human DHX36.13 To this end, we performed the FP-assay against FAM-labelled HRAS G4 (antiparallel),17 hTelo18 and BCL-219 G4s (mixed type). DM039 displayed negligible binding to either HRAS or hTelo G4s, confirming the inability of this peptide to bind some non-parallel G4-structures that were previously reported (see ESI†).13 On the other hand, the DNA G4 found in the promoter region of the BCL-2 oncogene, which also folds into a mixed-type conformation,19 was bound by DM039 with an observed Kd of 579 nM [447–749 nM – 95% CI]. Although the binding affinity is lower to what observed for MYC G4 (∼5 fold), this evidence suggested that DM039 could interact with some non-parallel G4s, possibly due to an unusually high accessibility of the G-tetrads in this mixed-type structure.
We next sought to confirm the high binding affinity displayed by DM039 to MYC G4 using Surface Plasmon Resonance (SPR) to measure the dissociation constant of DM039 bound to MYC G4. Strikingly, SPR data confirmed high binding affinity of DM039 to MYC G4, providing a measured Kd of 123 ± 23 nM (Fig. 3), which is in excellent agreement with the value obtained by FP.
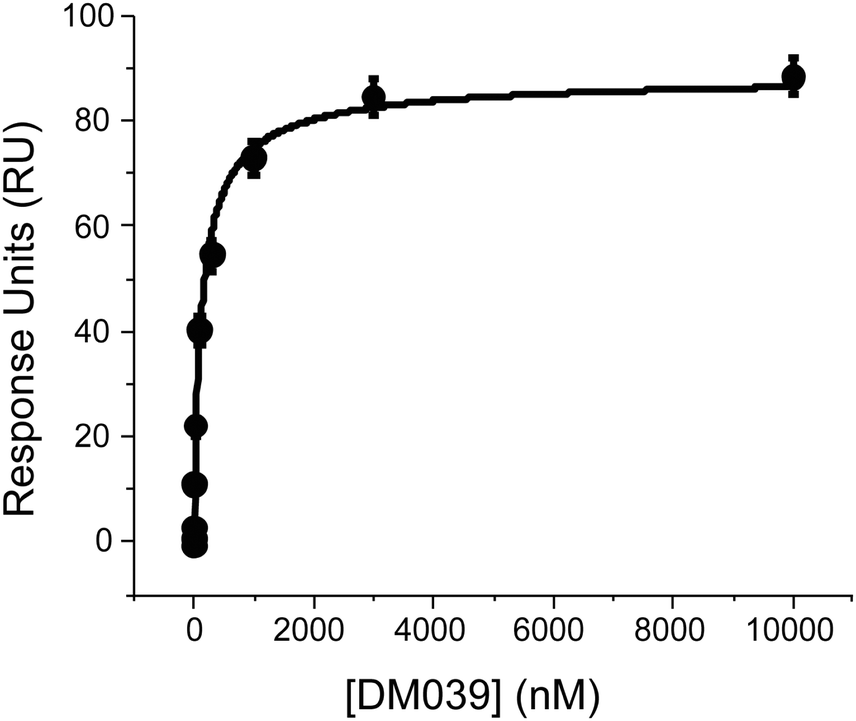 | ||
| Fig. 3 SPR experiments to study the interaction of DM039 and MYC G4 where the ligand concentration was varied from 0.1 nM to 10 μM. | ||
Altogether, our biophysical study revealed that the DM039 peptide preferentially binds MYC G4 with high selectivity over single-stranded and double-stranded DNA. Furthermore, DM039 displayed a significant degree of intra-G4 selectivity that, in contrast with previous reports, is not simply dictated by G4-topology but rather dependent on specific G4-structural features. This suggests that peptide-based binding of G4s might be the key to achieve individual G4-targeting by means of molecular probes.
The 22-amino acid sequence that constitutes DM039 is folded into a α-helix in the crystal structure reported of bovine DHX36 bound to MYC G4.11 To test whether DM039 also adopts a helical conformation when bound to MYC G4, we have performed CD analysis of DM039 in solution and in the presence of MYC G4 (see ESI†). Whilst DM039 appear to be unstructured in solution, an α-helical CD signal becomes detectable upon binding of the peptide to MYC G4, displaying an increase in the calculated helical fraction (fH) from 0.073 measured with DM039 in solution to 0.155 observed upon MYC G4 binding (see ESI†). Therefore, we wanted to investigate whether conformationally constraining the wild type peptide using a hydrocarbon staple could pay the entropic penalty of folding and improve the binding affinity of DM039 to MYC G4.
To test this hypothesis, we designed two stapled peptide analogues of the wild type peptide, DM083 (i, i + 4 type staple) and DM102 (i, i + 7 type staple) (Fig. 4A and B). On inspection of the helical interaction motif of bovine DHX36 bound to MYC G4 in the crystal structure and using the helix wheel, there are two ideal positions on the back face of the helix with which to incorporate a conformational constraint. The i, i + 4 stapled peptide DM083 was designed to incorporate the hydrocarbon bridge across one loop of the helix between residues K71 and Q75 (Fig. 4A). A longer i, i + 7 hydrocarbon constrain was also designed that constrains across two loops of the helix, between residues E64 and K71 (Fig. 4B). Importantly, the residues mutated do not appear to take part in the binding event with MYC G4 or intramolecular stabilising interactions. The linear peptides were synthesised using standard Fmoc/tBu SPPS conditions using a microwave assisted peptide synthesiser. Commercially available Fmoc-S5-OH and Fmoc-R8-OH were used to incorporate the nonproteinogenic amino acids with the appropriate alkene functionality required for ring-closing metathesis, which was achieved using Grubbs 1st Generation Catalyst. The resulting cyclised peptides were cleaved from solid support and globally deprotected under standard acidic conditions (TFA/TIS/H2O). The crude peptides were then purified by RP-HPLC and characterised using analytical RP-HPLC and mass spectrometry (ESI) (see ESI†).
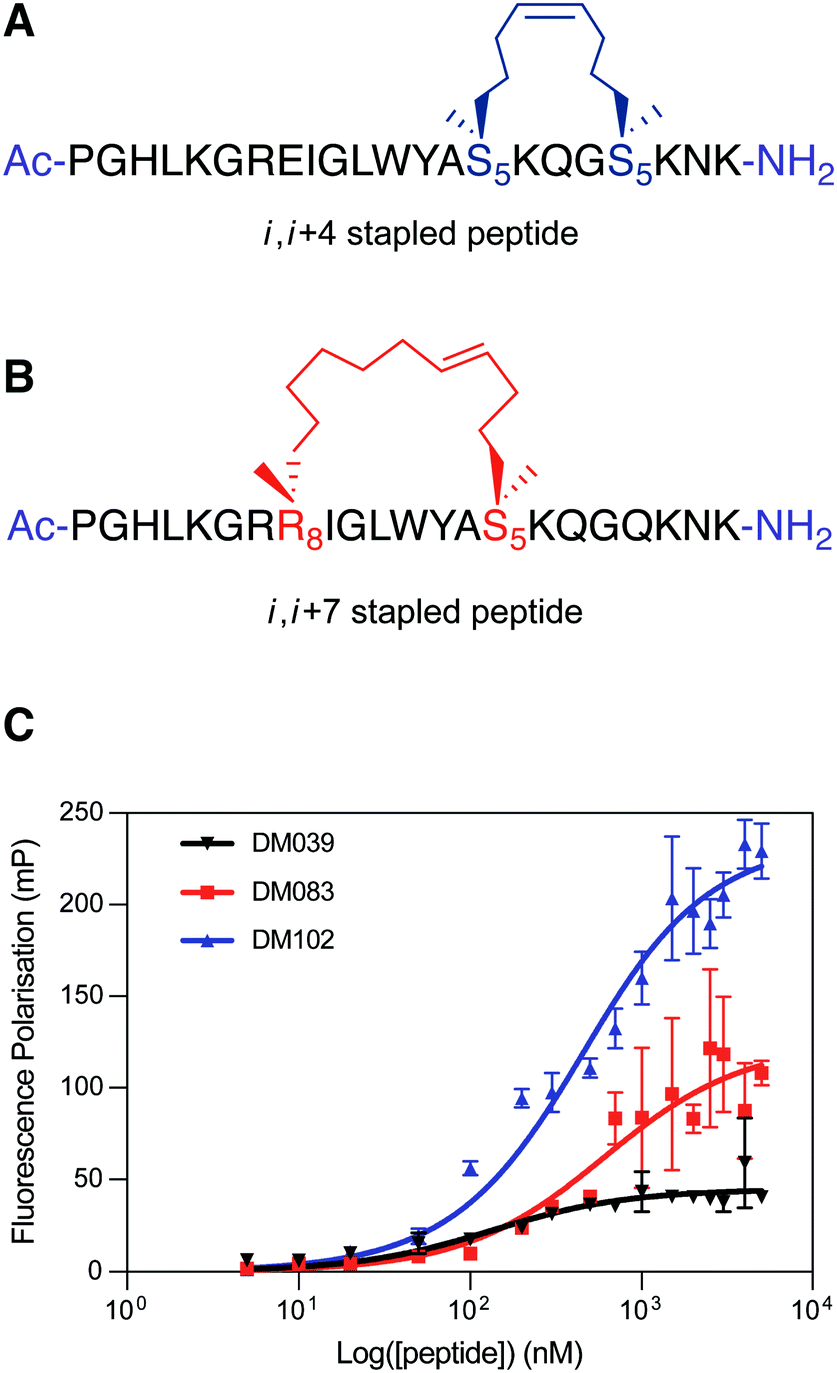 | ||
| Fig. 4 (A) i, i + 4 hydrocarbon stapled peptide DM083, (B) i, i + 7 hydrocarbon stapled peptide DM102. (C) Differential binding behaviour against MYC G4 observed for DM039 and its two stapled versions (DM083 and DM102) by means of FP measurements. The FP experiments were performed in Inner cell buffer (see ESI†), at room temperature, by using a fixed concentration of oligomer (20 nM) and increasing concentrations of peptide (0–5000 nM) (see ESI†). Each measurement was repeated three times. | ||
We first validated that stapling increased the helicity of the peptides by CD, observing a fH of 0.18 and 0.22 for DM083 and DM102 respectively (see ESI†). Crucially, the fH values of both DM083 and DM102 did not increase in the presence of MYC G4 (see ESI†). The two stapled peptides were then assessed for binding to MYC G4 by means of our FP-assay. Both DM083 and DM102 displayed a significant reduction of binding affinity to MYC G4, with measured Kd of 548 nM [317–934 nM – 95% CI] and 332 nM [249–435 nM – 95% CI] respectively (Fig. 4C). This suggested that stabilising the helical peptide conformation reduces the affinity for the targeted G4-structure (see ESI†). Whilst i, i + 4 stapling in DM083 reduces binding affinity to MYC G4 of ∼5 fold, i, i + 7 stapled peptide DM102 still retains substantial binding affinity to MYC G4 with a reduction of ∼3 fold compared to DM039. This is in agreement with a recent report that revealed how lactam stapling of a similar peptide sequence overall retained G4-binding ability.20 Nevertheless, we wanted to evaluate whether chemically locking of the peptide into a helical conformation, observed in the DHX36 crystal structure, resulted in retention of preferential selectivity for MYC G4, since the DHX36 helicase, unlike DM039, does not display intrinsic selectivity for a specific G4-structure or topology. We therefore tested the i, i + 7 stapled peptide DM102 for binding to parallel and non-parallel DNA G4s as well as to single- and double-stranded DNA controls. Surprisingly, we observed that DM102 binds with similar binding affinities all the oligonucleotides tested, yielding observed Kd values of 574 nM [381–855 nM – 95% CI] for c-KIT1, 1081 nM [847–1383 nM – 95% CI] for hTelo, 831 nM [622–1112 nM – 95% CI] for BCL-2, 1238 nM [827–1878 nM – 95% CI] for HRAS, 2628 nM [1648–4497 nM – 95% CI] for MYC-mutant and 2377 nM [1440–4222 nM – 95% CI] for double-stranded DNA. This observation suggested that DM039 binds to MYC G4 through an induced fit mechanism and a good degree of flexibility is required of the extended peptide backbone for both high affinity interaction and selectivity, as chemical stapling of this sequence in DM102 rendered the peptide a non-specific DNA binder.
In conclusion, we have developed a FP-assay to measure binding affinity of a short peptide (22 amino acids) extracted from a crystal structure of the bovine DHX36 helicase bound to MYC G4. Although selectivity towards parallel G4s was previously suggested for similar peptide sequences, we report that the particular peptide used in this work displays negligible binding to other parallel G4-structures (c-KIT1 and c-KIT2), binding to a mixed type G4 (BCL-2) and a preferential binding to MYC G4. Furthermore, we observed that chemical stapling of this peptide might preserve MYC binding but abrogates intra-G4 selectivity as well as selectivity over single and double-stranded DNA, suggesting that a rigid, extended peptide α-helical conformation is detrimental to the observed MYC selectivity. We anticipate that further investigation on DM039 binding modes and non-canonical chemical stapling will be key to develop a MYC-selective probe and disentangle the biological role of this particular G4 over the ∼700000 present in the human genome. Our findings might pave the way towards the rational design of peptide-based molecular probes for selective targeting of individual G4s.
Conflicts of interest
There are no conflicts to declare.References
- R. Hänsel-Hertsch, M. Di Antonio and S. Balasubramanian, Nat. Rev. Mol. Cell Biol., 2017, 18, 279–284 CrossRef PubMed.
- V. S. Chambers, G. Marsico, J. M. Boutell, M. Di Antonio, G. P. Smith and S. Balasubramanian, Nat. Biotechnol., 2015, 33, 877–883 CrossRef.
- S. Balasubramanian, L. H. Hurley and S. Neidle, Nat. Rev. Drug Discovery, 2011, 10, 261–275 CrossRef CAS PubMed.
- D. Rhodes and H. J. Lipps, Nucleic Acids Res., 2015, 43, 8627–8637 CrossRef CAS PubMed.
- K. Paeschke, M. L. Bochman, P. D. Garcia, P. Cejka, K. L. Friedman, S. C. Kowalczykowski and V. A. Zakian, Nature, 2013, 497, 458–462 CrossRef CAS PubMed.
- L. Froetscher, K. Marosi, D. M. Wilson, D. M. Eckley, S. Ghosh, M. P. Mattson, I. G. Goldberg, H. Kassahun, R. W. Maul, M. Gorospe, E. F. Fang, A. Tseng, S. K. Bharti, P. Bastian, M. Scheibye-Knudsen, V. A. Bohr, T. Iyama, S. De, M. Borch Jensen, R. M. Brosh, H. Nilsen and K. Scheibye-Alsing, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, 12502–12507 CrossRef.
- A. R. Haeusler, C. J. Donnelly, G. Periz, E. A. J. Simko, P. G. Shaw, M. S. Kim, N. J. Maragakis, J. C. Troncoso, A. Pandey, R. Sattler, J. D. Rothstein and J. Wang, Nature, 2014, 507, 195–200 CrossRef CAS.
- F. Raguseo, S. Chowdhury, A. Minard and M. Di Antonio, Chem. Commun., 2020, 56, 1317–1324 RSC.
- R. Hänsel-Hertsch, D. Beraldi, S. V. Lensing, G. Marsico, K. Zyner, A. Parry, M. Di Antonio, J. Pike, H. Kimura, M. Narita, D. Tannahill and S. Balasubramanian, Nat. Genet., 2016, 48, 1267–1272 CrossRef PubMed.
- D. Monchaud and M. P. Teulade-Fichou, Org. Biomol. Chem., 2008, 6, 627–636 RSC.
- M. C. Chen, R. Tippana, N. A. Demeshkina, P. Murat, S. Balasubramanian, S. Myong and A. R. Ferré-D’amaré, Nature, 2018, 558, 465–469 CrossRef CAS PubMed.
- The PyMOL Molecular Graphics System, Version 2.1.1.
- B. Heddi, V. V. Cheong, H. Martadinata and A. T. Phan, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 9609–9613 CrossRef PubMed.
- A. De Cian, L. Guittat, M. Kaiser, B. Saccà, S. Amrane, A. Bourdoncle, P. Alberti, M. P. Teulade-Fichou, L. Lacroix and J. L. Mergny, Methods, 2007, 42, 183–195 CrossRef CAS PubMed.
- N. J. Moerke, Curr. Protoc. Chem. Biol., 2009, 1, 1–15 CrossRef PubMed.
- S. Rankin, A. P. Reszka, J. Huppert, M. Zloh, G. N. Parkinson, A. K. Todd, S. Ladame, S. Balasubramanian and S. Neidle, J. Am. Chem. Soc., 2005, 127, 10584–10589 CrossRef CAS.
- A. Membrino, S. Cogoi, E. B. Pedersen and L. E. Xodo, PLoS One, 2011, 6, e24421 CrossRef CAS PubMed.
- A. Ambrus, D. Chen, J. Dai, T. Bialis, R. A. Jones and D. Yang, Nucleic Acids Res., 2006, 34, 2723–2735 CrossRef CAS PubMed.
- J. Dai, D. Chen, R. A. Jones, L. H. Hurley and D. Yang, Nucleic Acids Res., 2006, 34, 5133–5144 CrossRef CAS.
- M. Y. Yanevaa, V. V. Cheong, J. K. Cheng, K. W. Lim and A. T. Phan, Biochem. Biophys. Res. Commun., 2020 DOI:10.1016/j.bbrc.2020.02.144.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0cc02954h |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2020 |