
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence2-Mercaptobenzoxazoles: a class of carbonic anhydrase inhibitors with a novel binding mode to the enzyme active site†
Murat
Bozdag
 a,
Claudiu T.
Supuran
a,
Davide
Esposito
b,
Andrea
Angeli
a,
Fabrizio
Carta
a,
Simona Maria
Monti
b,
Giuseppina
De Simone
*b and
Vincenzo
Alterio
*b
a,
Claudiu T.
Supuran
a,
Davide
Esposito
b,
Andrea
Angeli
a,
Fabrizio
Carta
a,
Simona Maria
Monti
b,
Giuseppina
De Simone
*b and
Vincenzo
Alterio
*b
aNeurofarba Department, Section of Pharmaceutical and Nutriceutical Sciences, Università degli Studi di Firenze, Sesto Fiorentino, Florence, Italy
bIstituto di Biostrutture e Bioimmagini-CNR, Naples, Italy. E-mail: giuseppina.desimone@cnr.it; vincenzo.alterio@cnr.it; Tel: +39-081-2534579 Tel: +39-081-678674
First published on 23rd June 2020
Abstract
2-Mercaptobenzoxazole is a widely used organic scaffold in medicinal chemistry. By means of kinetic and structural studies, we demonstrate that this molecule can effectively be used to inhibit hCAs showing a peculiar binding mode. The results reported here can pave the way for the development of selective CA inhibitors.
Human carbonic anhydrases (hCAs) are zinc-containing enzymes that catalyze the reversible hydration of carbon dioxide to bicarbonate ion and proton.1 In humans, 12 catalytically active isoforms have been identified, which differ in tissue distribution, kinetic properties, oligomeric arrangement and sensitivity to inhibitors.1,2 Based on their sub-cellular localization, these isoforms can be classified into four different groups: cytosolic (CA I, II, III, VII, XIII), mitochondrial (CA VA and VB), membrane-associated (CA IV, IX, XII and XIV) and secreted (CA VI).1,2 A large number of structural studies showed that, as expected on the basis of their high sequence homology, these isoforms present a high structural similarity of the catalytic domain, which is characterized by a central twisted mainly antiparallel β-sheet surrounded by helical connections and additional β-strands. The active site is located in a wide and deep cavity, containing a zinc ion at the bottom, tetrahedrally coordinated by three conserved His residues and a water molecule/hydroxide ion,1 which is essential for the catalytic mechanism. A small pocket delimited by the conserved residues Val121, Val143, Leu198 and Trp209 within this cavity constitutes the CO2 binding site (Fig. S1, ESI†).
hCAs are involved in a wide range of physiological processes in many tissues and organs, such as pH regulation, gas exchange, ion transport, bone resorption, fatty acid metabolism, and many others; thus abnormal levels or activities of these enzymes are commonly associated with various diseases.1,2 For this reason, in recent years, CAs have become well-established drug targets and many CA inhibitors (CAIs), mostly belonging to the sulfonamide class, have been developed and clinically used for the management of a variety of disorders such as glaucoma and edema, or used as anticonvulsants, and antiobesity and antitumor drugs.1,2 However, since the currently clinically used CAIs generally do not show high selectivity for a specific isozyme,1,2 new molecules are constantly being investigated with the aim to develop isozyme-specific compounds devoid of undesired side effects. Carboxylates, phenols, carbamates, benzoxaboroles, polyamines, mercaptobenzothiazoles, sulfocoumarins and coumarins are only some examples of the huge number of new chemotypes that have been characterized as hCA inhibitors until now.1,3–7
Benzoxazole (Fig. 1) and its derivatives are important molecular scaffolds that play a central role in the drug discovery process thanks to their wide spectrum of pharmacological properties.8–11 Indeed, several drugs containing a benzoxazole core are actually available on the market. Benoxaprofen, boxazomycin B, calcimycin, chlorzoxazone and flunoxaprofen are some examples.12 Among the various benzoxazole derivatives, 2-mercaptobenzoxazoles (Fig. 1) constitute a very interesting compound family due to their applications not only in organic synthesis, but also in the biomedical field as antibacterial, antiviral, anti-inflammatory, analgesic and anthelmintic agents.13–17 Moreover, interesting studies have shown that the complexes of 2-mercaptobenzoxazoles with various transition metals possess a cytotoxic activity against cancer cells.18
 | ||
| Fig. 1 Chemical structures of benzoxazole, benzensulfonamide (BNZ), 2-mercaptobenzoxazole (1) and its sulfonamide derivative (2). | ||
Recently, some aminomethyl and alkoxymethyl 2-mercaptobenzoxazole derivatives have been investigated for their inhibition properties against hCAs. These compounds, found to be potent inhibitors of isoforms I and II, emerged as new and interesting candidates for the development of drugs towards CA-related diseases.7 However, almost no information is currently available on their inhibition mechanism. To get more insights into the CA inhibition properties of this class of molecules, in the present study, we report the synthesis and the kinetic characterization of 2-mercaptobenzoxazole (1), as well as the crystallographic structure of the complex that this molecule forms with hCA II. The comparison with its sulfonamide derivative 2 (Fig. 1) is also described, providing interesting perspectives on the utilization of 2-mercaptobenzoxazoles as selective CAIs.
2-Mercaptobenzoxazole 1 was obtained by a single step synthesis treating 2-aminophenol with thiophosgene at 0 °C in dry tetrahydrofuran.19,20 A stopped-flow CO2 hydratase assay21 was used to investigate the inhibition activity of this compound against the human CA isoforms of particular relevance such as I, II, IX and XII. The obtained data were compared with the reference inhibitors benzensulfonamide (BNZ) and acetazolamide (AAZ) and are reported in Table 1.
| K I (μM) | ||||
|---|---|---|---|---|
| Cmp. | hCA I | hCA II | hCA IX | hCA XII |
| a Mean from 3 different assays, by a stopped flow technique (errors were in the range of ±5–10% of the reported values). b Inhibition values @6 h incubation. c Inhibition values @15 min incubation. d Data were taken from Di Fiore et al.22 | ||||
| 1 | 88.4 | 0.97 | 20.7 | 1.9 |
| 2 | 2.9 | 0.48 | 0.03 | 0.007 |
| BNZ | 0.086 | 0.101 | 0.097 | 0.090 |
| AAZ | 0.25 | 0.012 | 0.026 | 0.006 |
2-Mercaptobenzoxazole 1 inhibited hCA isoforms in the range of 0.97–88.4 μM after at least 6 hours incubation, in agreement with the kinetic trend of aryl thiol containing compounds previously reported.1,7,23 Compound 1 showed preferential inhibition of the hCA II isoform with a KI of 0.97 μM, followed by hCA XII (KI 1.9 μM). Of interest are the selectivity indexes (SIs) of 1 within the cytosolic and the tumor related hCA isoforms. The SI of the former (SIcyt: KI hCA I/KI hCA II) is 91.1 and of the latter (SItum: KI hCA IX/KI hCA XII), it is 10.9. Such results are quite indicative of the high degree of hCA isoform selectivity of the mercaptobenzoxazole scaffold, which therefore may be considered for further biomedical development.
To elucidate the binding mode of 2-mercaptobenzoxazoles to the CA active site, the crystallographic structure of the adduct formed by compound 1 with the best inhibited isoform (hCA II) was determined. Crystals of the complex were obtained by soaking techniques and were isomorphous with those of the native protein,24 allowing for the analysis of the three-dimensional structure by difference Fourier techniques. Data collection and refinement statistics are reported in Table S1 (ESI†).
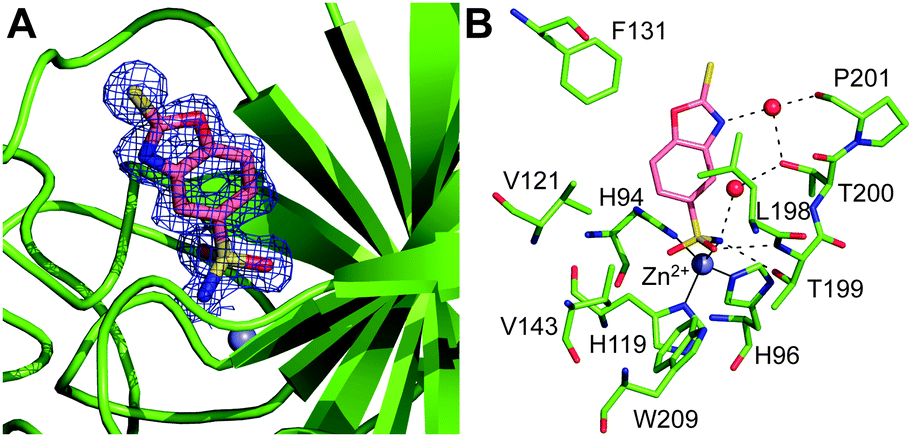
Inspection of the electron density maps at several stages of refinement clearly indicated the binding of one inhibitor molecule within the hCA II active site cavity (Fig. 2A). The binding of this molecule did not generate significant changes in the hCA II structure. Indeed, the r.m.s.d. for the superposition of the corresponding Cα atoms between the native enzyme and the enzyme-inhibitor complex was 0.3 Å.
 | ||
| Fig. 2 (A) σA-weighted (|2Fo–Fc|, ϕc) simulated annealing omit map (contoured at 1.0σ) relative to the inhibitor molecule in the hCA II/1 complex. (B) Details of the interactions of 1 with the enzyme active site. Residues involved in polar and hydrophobic interactions are shown. Continuous lines indicate zinc ion coordination and dashed lines indicate H-bond distances. | ||
2-Mercaptobenzoxazoles are well known for their ability to bind metal ions,25,26 using various binding modes: either a monodentate coordination through the exocyclic sulfur or the endocyclic nitrogen atom, or bidentate coordination through both sulfur and oxygen/nitrogen atoms.27 In the hCA II/1 adduct, a monodentate coordination of the inhibitor was observed. Indeed, the 1 sulfur atom binds to the metal ion, changing its coordination from the tetrahedral one, observed in the native enzyme, to a distorted trigonal bipyramidal one (Fig. 2B). The two axial positions of the bipyramid were occupied by His96 and the sulfur atom of 1, while the three equatorial positions were occupied by His94, His119 and the zinc-bound solvent molecule, which, as a consequence of the inhibitor binding, slightly moved from its canonical position24 (Fig. S2, ESI†). Surprisingly, the Zn–S distance (2.75 Å) was longer than what was expected based on the sum of the covalent radii (2.33 Å).28,29 However, an extensive analysis of Zn–S coordination distance in the Cambridge Structural Database30 showed the presence of several pentacoordinated zinc complexes with different sulfur donor ligands, characterized by a Zn–S distance even longer than 2.75 Å.29,31,32
It is worth noting that the inhibitor sulfur atom is located approximately in the same position occupied by CO2 in the hCA II/substrate complex33 (see Fig. S3, ESI†). Compound 1 also established a hydrogen bond interaction between its endocyclic oxygen and the zinc-bound solvent molecule (Fig. 2B), which was assumed to be a water molecule in agreement with the hCA II neutron structure solved at pH 9.34 Both the coordination to the zinc ion and the hydrogen bond to the zinc-bound water molecule have been extensively described as a way to anchor inhibitors to the CA active site.6,35,36 To date, only a few inhibitors, mainly very small molecules such as acetate, perchlorate and thiocyanate35,37,38 (Fig. S4, ESI†), have been shown to adopt simultaneously both binding modes. Thus, this is the first evidence of such an event for a more complex molecule. No other hydrogen bond interaction was observed between the enzyme and the inhibitor molecule, but some van der Waals contacts with residues Gln92, His94, His119, Val121, Phe131, Leu198, Thr199, Thr200 and Trp209 were observed (Fig. 2B). Interestingly, the sequence alignment between the investigated hCAs (see Fig. S5, ESI†) revealed that protein residues in the hCA II/1 complex that interact with the inhibitor are not all conserved in the other isoforms, thus suggesting possible modifications in the binding of 1 to the diverse isoforms and explaining the differences observed in the inhibition constants (see Table 1).
The structural superposition between the hCA II/1 and hCA II/BNZ39 adducts showed that, despite the very different chemical structure of these two molecules and their diverse binding modes to the metal ion (trigonal bipyramidal for the first and tetrahedral for the second), their organic scaffold occupies a similar position within the enzyme active site (Fig. 3). These results suggest that, as largely described for benezenesulfonamides,1 the aromatic ring of 2-mercaptobenzoxazoles can be derivatized to establish further interactions with the less conserved regions of the CA catalytic cavity.
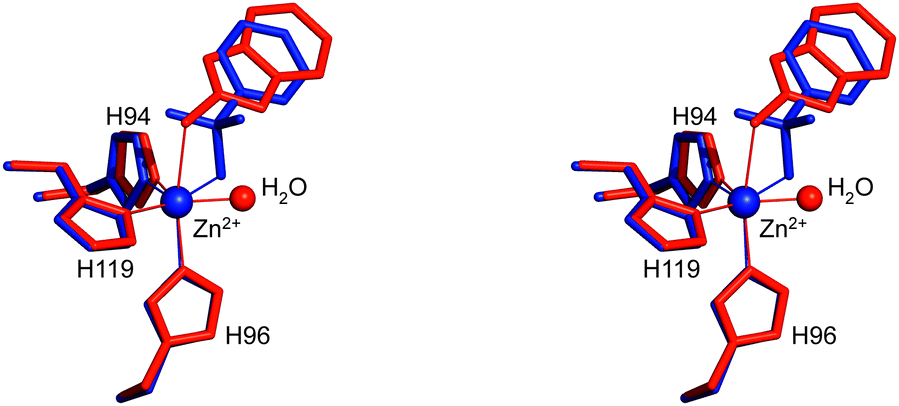 | ||
| Fig. 3 Stereo view of the structural superposition between compound 1 (red) and BNZ (blue, PDB code 2WEJ), when bound to the hCA II active site.39 Continuous lines indicate Zn coordination. | ||
To further investigate the different inhibition behaviour of the mercaptobenzoxazole moiety and the classical primary sulphonamide, a derivative containing a sulfonamide moiety in position 5 of the benzoxazole ring (see compound 2 in Fig. 1) was synthesized, tested as an inhibitor and characterized by X-ray crystallography. Synthesis was carried out as described for compound 1, using 2-aminophenol-4-sulfonamide as a starting compound.19,20 Again, a stopped-flow CO2 hydratase assay was used to investigate the inhibitory properties of this compound, showing that the introduction of the canonical sulfonamide metal binding moiety1 determined a radical change in the kinetic profile. Indeed, compound 2 showed its inhibition potency after 15 min incubation, thus suggesting that the sulfonamide moiety was the real metal coordination species, ruling out the thiol as a competing moiety.1,7,23 As reported in Table 1, compound 2 poorly inhibited the cytosolic isoforms hCA I and II (KIs of 2.9 and 0.48 μM, respectively), whereas it effectively inhibited hCA IX and XII (KIs of 30 and 7 nM, respectively). Interestingly, although the inhibition potencies of 2 against the isoforms here considered were generally improved in comparison to 1 (see Table 1), the calculated SIcyt and SItum were heavily reduced (6.0 and 4.3, respectively).
Similar to the hCA II/1 complex, the crystals of the hCA II/2 adduct were obtained by soaking techniques. In Table S1 (ESI†) are summarized all the statistics for data collection and refinement. Different from the hCA II/1 complex, in this case, the analysis of the electron density maps revealed the binding to the enzyme of two inhibitor molecules: one in the active site cavity (Fig. 4A) and the other on the protein surface, in a cleft already described for other hCA II/inhibitor complexes40 (see Fig. S6, ESI†). The binding of this second inhibitor molecule will not be discussed here, since it occurs far from the active site, thus it is not correlated with the inhibition properties of the molecule. In contrast, the binding of the molecule in the active site will be analysed in detail since it is clearly associated with the inhibitory potency of the investigated compound. As described for the hCA II/1 complex, also in this case the binding of the inhibitor to hCA II did not generate significant changes in the enzyme structure (r.m.s.d. for the superposition of hCA II Cα atoms in the bound and unbound form is 0.3 Å).
 | ||
| Fig. 4 (A) σA-weighted (|2Fo–Fc|, ϕc) simulated annealing omit map (contoured at 1.0σ) relative to the inhibitor molecule in the hCA II/2 complex. (B) Details of the interactions of 2 with the hCA II active site. Residues involved in polar and hydrophobic interactions are shown. Continuous lines indicate zinc ion coordination, whereas dashed lines indicate H-bond distances. | ||
Interestingly, 2 showed a binding mode to the enzyme completely different with respect to 1. Indeed, in this case, the sulfonamide moiety coordinates the catalytic zinc ion, establishing the canonical interactions observed in other CA/sulfonamide adducts1 (see Fig. 4B). Consequently, the 2-mercaptobenzoxazole ring is rotated by about 180° with respect to 1, with the exocyclic sulfur pointing toward the exit of the active site cavity (Fig. S7, ESI†). Two water-mediated hydrogen bond interactions are present between the inhibitor and Thr200OG1 and Pro201O (Fig. 4B). Several van der Waals contacts between the bicyclic ring and residues Gln92, His94, Val121, Phe131, Leu198, Thr200 and Pro201 further stabilize the binding. It is worth noting that, as also suggested by our kinetic data, although containing two zinc binding moieties, the anchoring to the metal ion of compound 2 is mediated only by the sulfonamide, clearly indicating that sulfonamide is a zinc binding group much stronger than the sulphur atom of 2-mercaptobenzoxazole. This is a very interesting finding since benzenesulfonamides, although very potent inhibitors, are generally poorly selective against the different CA isoforms. A huge number of papers have reported the introduction on the phenyl ring of substituents that, interacting with the less conserved regions of the CA active site, should induce selectivity. However, very often, due to the dominant contribution of the sulfonamide moiety in the interaction with the enzyme, the presence of these substituents has only a marginal effect on the enzyme-inhibitor affinity, thus not allowing the discrimination between the different isoforms.22,41 In the case of 2-mercaptobenzoxazoles, the presence of a less strong zinc binding moiety could cause a greater influence of the ring substituents on the inhibitor binding affinity and consequently could allow the obtainment of more selective CAIs.
In conclusion, our results clearly indicate that 2-mercaptobenzoxazole represents an interesting lead compound alternative to the classical sulfonamides for the development of selective CAIs. Even if the absence of the sulfonamide moiety on one side reduces the affinity for the enzyme, on the other side, it opens the possibility to finely modulate the selectivity for a specific CA isozyme. Indeed, by using the so-called tail approach, largely applied for benzensulfonamide inhibitors,1 different chemical groups can be introduced on the aromatic ring, allowing the targeting of the most variable regions in the active site of the various CA isozymes.
This work was supported by a grant from MIUR-PON “Ricerca e Innovazione” 2014–2020 (grant MOLIM ONCOBRAIN LAB) and a grant from Regione Campania PO FESR 2014–2020 (grant eMORFORAD).
Conflicts of interest
There are no conflicts to declare.Notes and references
- V. Alterio, A. Di Fiore, K. D'Ambrosio, C. T. Supuran and G. De Simone, Chem. Rev., 2012, 112, 4421–4468 CrossRef CAS PubMed.
- C. T. Supuran, Nat. Rev. Drug. Discovery, 2008, 7, 168–181 CrossRef CAS PubMed.
- V. Alterio, R. Cadoni, D. Esposito, D. Vullo, A. Di Fiore, S. M. Monti, A. Caporale, M. Ruvo, M. Sechi, P. Dumy, C. T. Supuran, G. De Simone and J. Y. Winum, Chem. Commun., 2016, 52, 11983–11986 RSC.
- G. De Simone, A. Angeli, M. Bozdag, C. T. Supuran, J. Y. Winum, S. M. Monti and V. Alterio, Chem. Commun., 2018, 54, 10312–10315 RSC.
- K. Tars, D. Vullo, A. Kazaks, J. Leitans, A. Lends, A. Grandane, R. Zalubovskis, A. Scozzafava and C. T. Supuran, J. Med. Chem., 2013, 56, 293–300 CrossRef CAS PubMed.
- A. Karioti, F. Carta and C. T. Supuran, Molecules, 2016, 21, 1649 CrossRef CAS PubMed.
- I. Gulcin, M. Abbasova, P. Taslimi, Z. Huyut, L. Safarova, A. Sujayev, V. Farzaliyev, S. Beydemir, S. H. Alwasel and C. T. Supuran, J. Enzyme Inhib. Med. Chem., 2017, 32, 1174–1182 CrossRef CAS PubMed.
- P. Malapati, V. S. Krishna, R. Nallangi, R. R. Srilakshmi and D. Sriram, Eur. J. Med. Chem., 2018, 145, 23–34 CrossRef CAS PubMed.
- C. K. Ryu, R. Y. Lee, N. Y. Kim, Y. H. Kim and A. L. Song, Bioorg. Med. Chem. Lett., 2009, 19, 5924–5926 CrossRef CAS PubMed.
- S. Aiello, G. Wells, E. L. Stone, H. Kadri, R. Bazzi, D. R. Bell, M. F. Stevens, C. S. Matthews, T. D. Bradshaw and A. D. Westwell, J. Med. Chem., 2008, 51, 5135–5139 CrossRef CAS PubMed.
- S. M. Sondhi, N. Singh, A. Kumar, O. Lozach and L. Meijer, Bioorg. Med. Chem., 2006, 14, 3758–3765 CrossRef CAS PubMed.
- S. Kakkar, S. Tahlan, S. M. Lim, K. Ramasamy, V. Mani, S. A. A. Shah and B. Narasimhan, Chem. Cent. J., 2018, 12, 92 CrossRef CAS PubMed.
- Y. Xiao, B. Jing, X. Liu, H. Xue and Y. Liu, Beilstein J. Org. Chem., 2019, 15, 279–284 CrossRef CAS PubMed.
- T. Tankam, J. Srisa, M. Sukwattanasinitt and S. Wacharasindhu, J. Org. Chem., 2018, 83, 11936–11943 CrossRef CAS PubMed.
- C. Safak, R. Simsek, K. Erol and K. Vural, Pharmazie, 1996, 51, 180–182 CAS.
- P. Kohli, S. D. Srivastava and S. K. Srivastava, J. Chin. Chem. Soc., 2013, 54, 8 Search PubMed.
- J. Mentado, H. Flores and P. Amador, J. Chem. Thermodyn., 2008, 40, 4 CrossRef.
- R. Mitra and A. G. Samuelson, Eur. J. Inorg. Chem., 2014, 11 Search PubMed.
- C. Ding, S. Wang, Y. Sheng, Q. Dai, Y. Zhao, G. Liang and Z. Song, RSC Adv., 2019, 9, 5 Search PubMed.
- R. E. Martin, L. G. Green, W. Guba, N. Kratochwil and A. Christ, J. Med. Chem., 2007, 50, 6291–6294 CrossRef CAS PubMed.
- R. G. Khalifah, J. Biol. Chem., 1971, 246, 2561–2573 CAS.
- A. Di Fiore, A. Maresca, V. Alterio, C. T. Supuran and G. De Simone, Chem. Commun., 2011, 47, 11636–11638 RSC.
- G. L. Almajan, A. Innocenti, L. Puccetti, G. Manole, S. Barbuceanu, I. Saramet, A. Scozzafava and C. T. Supuran, Bioorg. Med. Chem. Lett., 2005, 15, 2347–2352 CrossRef CAS PubMed.
- A. E. Eriksson, T. A. Jones and A. Liljas, Proteins, 1988, 4, 274–282 CrossRef CAS PubMed.
- T. Nakahodo, E. Horn and E. R. Tiekink, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 2000, 56(Pt 11), 1316–1318 CrossRef PubMed.
- R. Mitra, S. Das, S. V. Shinde, S. Sinha, K. Somasundaram and A. G. Samuelson, Chemistry, 2012, 18, 12278–12291 CrossRef CAS PubMed.
- J. Susperregui, M. Bayle, J. M. Léger and G. Déléris, J. Organomet. Chem., 1998, 556, 6 CrossRef.
- J. E. Huheey, Inorganic chemistry: principles of structure and reactivity, Harper & Row, New York, 2nd edn, 1978 Search PubMed.
- R. Baggio, A. Frigerio, E. B. Halac, D. Vega and M. Perec, J. Chem. Soc., Dalton Trans., 1992, 1887–1892 RSC.
- F. H. Allen, Acta Crystallogr., Sect. B: Struct. Sci., 2002, 58, 380–388 CrossRef PubMed.
- D. Chen, C. S. Lai and E. R. T. Tiekink, CrystEngComm, 2006, 8, 8 RSC.
- S. Thirumaran, V. Venkatachalam, A. Manohar, K. Ramalingam, G. Bocelli and A. Cantoni, J. Coord. Chem., 1998, 44, 2801–2808 CrossRef.
- B. Sjoblom, M. Polentarutti and K. Djinovic-Carugo, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 10609–10613 CrossRef PubMed.
- S. Z. Fisher, A. Y. Kovalevsky, J. F. Domsic, M. Mustyakimov, R. McKenna, D. N. Silverman and P. A. Langan, Biochemistry, 2010, 49, 415–421 CrossRef CAS PubMed.
- A. E. Eriksson, P. M. Kylsten, T. A. Jones and A. Liljas, Proteins, 1988, 4, 283–293 CrossRef CAS PubMed.
- F. Carta, C. Temperini, A. Innocenti, A. Scozzafava, K. Kaila and C. T. Supuran, J. Med. Chem., 2010, 53, 5511–5522 CrossRef CAS PubMed.
- J. M. Fox, K. Kang, W. Sherman, A. Heroux, G. M. Sastry, M. Baghbanzadeh, M. R. Lockett and G. M. Whitesides, J. Am. Chem. Soc., 2015, 137, 3859–3866 CrossRef CAS PubMed.
- A. Di Fiore, S. M. Monti, M. Hilvo, S. Parkkila, V. Romano, A. Scaloni, C. Pedone, A. Scozzafava, C. T. Supuran and G. De Simone, Proteins, 2009, 74, 164–175 CrossRef CAS PubMed.
- A. D. Scott, C. Phillips, A. Alex, M. Flocco, A. Bent, A. Randall, R. O'Brien, L. Damian and L. H. Jones, ChemMedChem, 2009, 4, 1985–1989 CrossRef CAS PubMed.
- G. De Simone, G. Pizika, S. M. Monti, A. Di Fiore, J. Ivanova, I. Vozny, P. Trapencieris, R. Zalubovskis, C. T. Supuran and V. Alterio, BioMed Res. Int., 2014, 2014, 523210 Search PubMed.
- A. Di Fiore, A. Maresca, C. T. Supuran and G. De Simone, Chem. Commun., 2012, 48, 8838–8840 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details, Fig. S1–S8 and Table S1. See DOI: 10.1039/d0cc02857f |
| This journal is © The Royal Society of Chemistry 2020 |