
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAsymmetric [N–I–N]+ halonium complexes†
Jas S.
Ward
 *a,
Giorgia
Fiorini
a,
Antonio
Frontera
b and
Kari
Rissanen
*a
*a,
Giorgia
Fiorini
a,
Antonio
Frontera
b and
Kari
Rissanen
*a
aUniversity of Jyvaskyla, Department of Chemistry, Jyväskylä 40014, Finland. E-mail: james.s.ward@jyu.fi; kari.t.rissanen@jyu.fi
bDepartment of Chemistry, Universitat de les Illes Balears, Crts de Valldemossa km 7.6, 07122 Palma de Mallorca (Baleares), Spain
First published on 20th June 2020
Abstract
The first asymmetric halogen-bonded iodonium complexes [I(py)(4-DMAP)]PF6 (2c) and [I(py)(4-Etpy)]PF6 (2e) were prepared via [N–Ag–N]+ → [N–I–N]+ cation exchange of their analogous 2-coordinate silver complexes. The complexes were characterised by 1H and 1H–15N HMBC NMR spectroscopy, and single crystal X-ray crystallography.
The popularisation of halogen bonding (XB) two decades ago has quickly matured into one of the most studied non-covalent interactions, even more so since 2007.1,2 The halogen bond, which was recently defined,3 occurs between the positive regions of the electrostatic potential surface of halogen atoms, acting as electrophilic XB donors, and neutral or anionic Lewis bases, as XB acceptors. Halogen bonding has rapidly found a niche in crystal engineering due to its directionality, specificity, and high strength, all of which combine to facilitate the synthesis of complex structures with a high degree of accuracy and precision.1,2,4
Polarisation of the halogen beyond its limit, whereupon the electron is removed from the halogen atom, generates a positively charged ion, i.e., the halonium ion X+ (X = Cl, Br, or I). Halonium ions can therefore be viewed as extremely polarised halogen atoms, and under the larger umbrella of halogen bonding as strong XB donors, recently reviewed by Erdélyi.5 However, the reactivity of halonium ions very much differentiates them from other classical halogen bond donors.
After their debut in the 1960's,6,7 halonium ions have found great utility in organic transformations including the electrophilic iodonation of unactivated arenes,8 the promotion of C–C and C–X bond formation,9 and the selective direct iodonation of peptides.10 This myriad of uses as a mild iodonating reagent is owed largely to the efforts of José Barluenga in popularising the now ubiquitous, and commercially available, Barluenga's reagent [I(py)2]BF4. Similarly, the effectiveness of asymmetric reagents in producing asymmetrically pure compounds has been realised and research into new variations is rampant in the literature.11,12 Therefore, the potential of asymmetric halonium reagents is a tempting target for modern synthetic chemists to explore. However, to date all reported halonium complexes (and even linear [AgL2]+ analogues) have involved a pair of homoleptic ligands,13–17 with no unrestrained heteroleptic examples being described, though ligand enforced examples have been previously observed in solution.18 Herein we report the first synthesis and characterisation of unrestrained heteroleptic (and therefore asymmetric) Ag+ and I+ complexes.
The asymmetric silver complexes were prepared by the 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1:1 combination of py:L:AgPF6 (py = pyridine; L = N,N-dimethylpyridin-4-amine (4-DMAP) or 4-ethylpyridine (4-Etpy)) in CH2Cl2, which resulted in the complexes [Ag(py)(4-DMAP)]PF6 (1c) and [Ag(py)(4-Etpy)]PF6 (1e) in 87 and 89% yields, respectively (Scheme 1). The 1H NMR spectra showed resonances consistent with the presence of a pyridine and either 4-DMAP or 4-Etpy, and more importantly, in a 1:1 py:L ratio.
:1:1 combination of py:L:AgPF6 (py = pyridine; L = N,N-dimethylpyridin-4-amine (4-DMAP) or 4-ethylpyridine (4-Etpy)) in CH2Cl2, which resulted in the complexes [Ag(py)(4-DMAP)]PF6 (1c) and [Ag(py)(4-Etpy)]PF6 (1e) in 87 and 89% yields, respectively (Scheme 1). The 1H NMR spectra showed resonances consistent with the presence of a pyridine and either 4-DMAP or 4-Etpy, and more importantly, in a 1:1 py:L ratio.
 | ||
| Scheme 1 The synthetic route to asymmetric Ag+ and I+ complexes 1 and 2. | ||
The asymmetric iodonium complexes [I(py)(4-DMAP)]PF6 (2c) and [I(py)(4-Etpy)]PF6 (2e) were prepared from their respective silver complexes via [N–Ag–N]+ to [N–I–N]+ cation exchange upon reaction with elemental iodine.17,19–25 Unlike their precursor silver complexes these reactions did not proceed cleanly, and resulted in mixtures of the desired asymmetric complexes in combination with the possible homoleptic products. The mechanism by which the iodonium-forming cation exchange proceeds has to date not been definitively established, however, this observed ligand scrambling during the process suggests that at some point during the reaction the ligands are at best only weakly coordinated to the Ag+/I+ centres, permitting their liberation and rearrangement to form the homoleptic complexes.
The 1H NMR analysis of the asymmetric silver complexes 1c and 1e were clean single species spectra, but the corresponding iodonium complexes always showed mixtures (Fig. 1), even if measured from the single crystal samples. This is probably due to fast exchange on the NMR time scale, the known high reactivity, and the ease of scrambling of the I+ complexes. The identity of both asymmetric silver and iodonium complexes was further confirmed by comparison to pure samples of the independently prepared symmetric complexes [Ag(py)2]PF6 (1a), [I(py)2]PF6 (2a), [Ag(4-DMAP)2]PF6 (1b), [I(4-DMAP)2]PF6 (2b),‡ [Ag(4-Etpy)2]PF6 (1d), and [I(4-Etpy)2]PF6 (2d) (see ESI†).
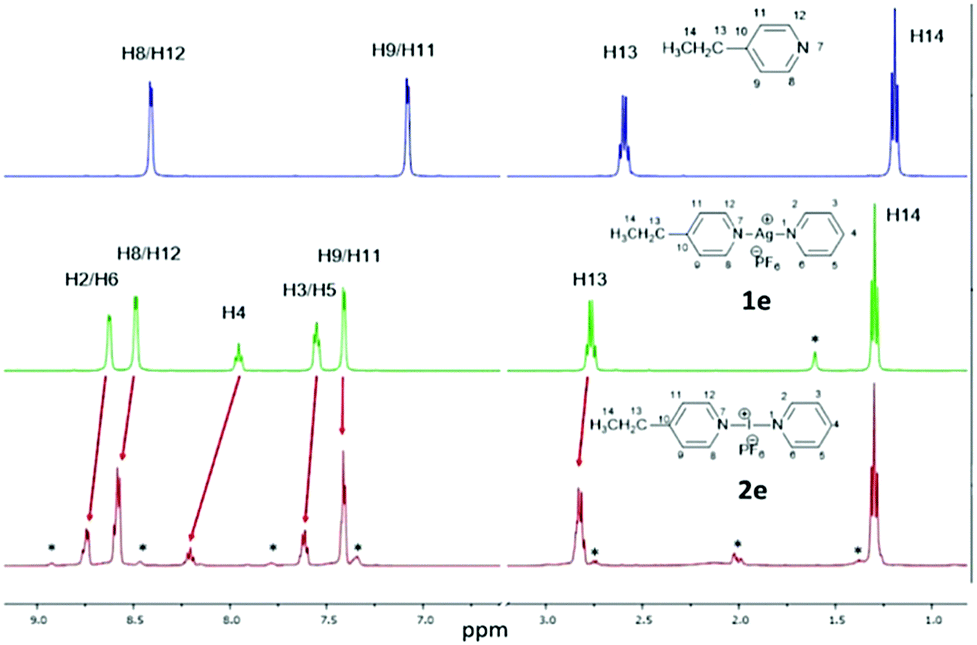 | ||
| Fig. 1 A comparison of the aromatic and alkyl regions of the 1H NMR spectra of the uncomplexed ligand (4-Etpy; top), the silver complex [Ag(py)(4-Etpy)]PF6 (1e; middle), and the iodonium complex [I(py)(4-Etpy)]PF6 (2e; bottom) in CD2Cl2 (the asterisks denote minor impurities or other unidentified species). | ||
Additional confirmation was obtained from 1H–15N HMBC NMR spectroscopy (Fig. 2 and ESI†). The chemical environment of the 15N nuclei were, as expected, found to be far more sensitive to the changes in complexation, making it a useful tool to distinguish the asymmetric complexes, especially of the asymmetric iodonium complexes that were observed as a varying mixture of [L1–I–L2]+, [L1–I–L1]+ and [L2–I–L2]+. Even if it cannot be excluded that complexes 2c and 2e exist as statistical mixtures of the possible respective symmetric (2a, 2b, 2d) and asymmetric forms (2c, 2e) in solution, the 15N NMR coordination shifts observed for the asymmetric 2c and 2e by 1H–15N HMBC are comparable to those reported for the analogous ligand enforced asymmetric complex synthesised by Erdélyi and co-workers,18 which is present in solution as a single asymmetric complex due to entropic reasons. Chemical shift changes of the coordinated aromatic nitrogen atoms when going from the uncomplexed ligands to the Ag+ complexes were in the range of 44–61 ppm, and within the range of 48–64 ppm upon conversion of the Ag+ to I+ complexes, making a total chemical shift change between 93–125 ppm. The smallest change in chemical shift was observed for the conversion of 1b to 2b (Δ 48.0 ppm), and the largest being for the 4-DMAP ligand of asymmetric 1c when reacted to 2c (Δ 63.9 ppm). Between the asymmetric complexes and their analogous symmetric reference complexes the chemical shift changes were within a much narrower range, with a range of 9–12 ppm observed for the conversion of the uncomplexed ligands to the Ag+ complexes, and between 3–20 ppm for the Ag+ to I+ conversions. These small but distinct chemical shift changes make this technique a convenient method to monitor whether the cation exchange process has proceeded, and will undoubtedly find increased utility in this regard in the future.
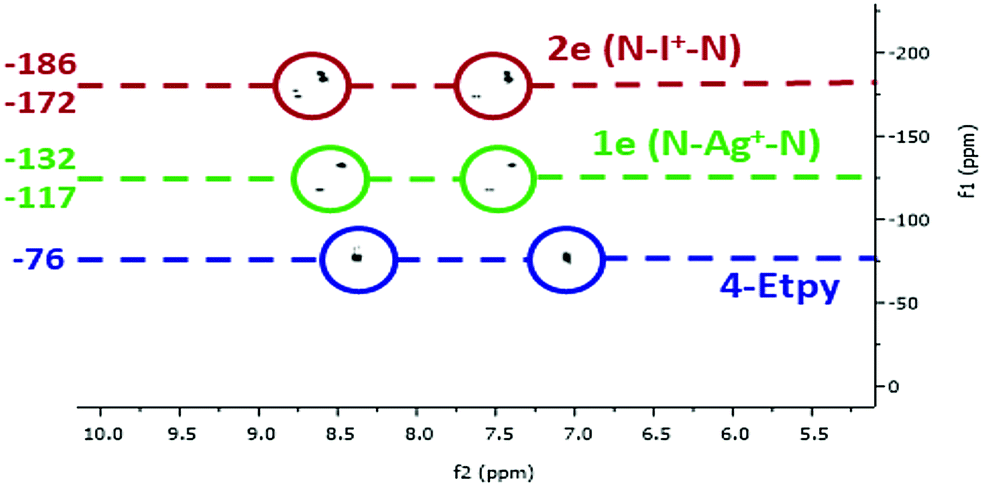 | ||
| Fig. 2 The comparison of the 1H–15N HMBC correlations of the uncomplexed ligand (4-Etpy), the silver complex [Ag(py)(4-Etpy)]PF6 (1e), and the iodonium complex [I(py)(4-Etpy)]PF6 (2e). All values in ppm and referenced to CD2Cl2 (1H) or CD3NO2 (15N). | ||
Due to their inherent reactivity (as halogenating reagents), to date there are only a relatively small number of halonium complexes characterised in the solid state, and all those complexes are symmetric, viz. [L–X–L]+, in nature. The asymmetric complexes 1c, 1e and 2c were also studied by single crystal X-ray crystallography (Fig. 3). The structure of 1c and 1e reveal asymmetry in the Ag+–N bond lengths with Ag–py/Ag–L bond lengths of 2.117(8)/2.121(9) Å for 1c and 2.131(7)/2.102(6) Å for 1e, respectively. Weak argentophilic Ag+⋯Ag+ interactions between adjacent complexes were observed for both 1c (3.484(2) Å) and 1e (3.3593(8) Å, see ESI†). The X-ray crystallographic study of 2c represents the first example of an asymmetric halonium complex. Complex 2c displays similar asymmetry of the I+–N bond lengths between the iodonium and the stronger XB acceptor (2.27(1) Å to 4-DMAP) in comparison to the weaker Lewis base (2.29(1) Å to py), as was previously discussed for its silver precursor 1c.
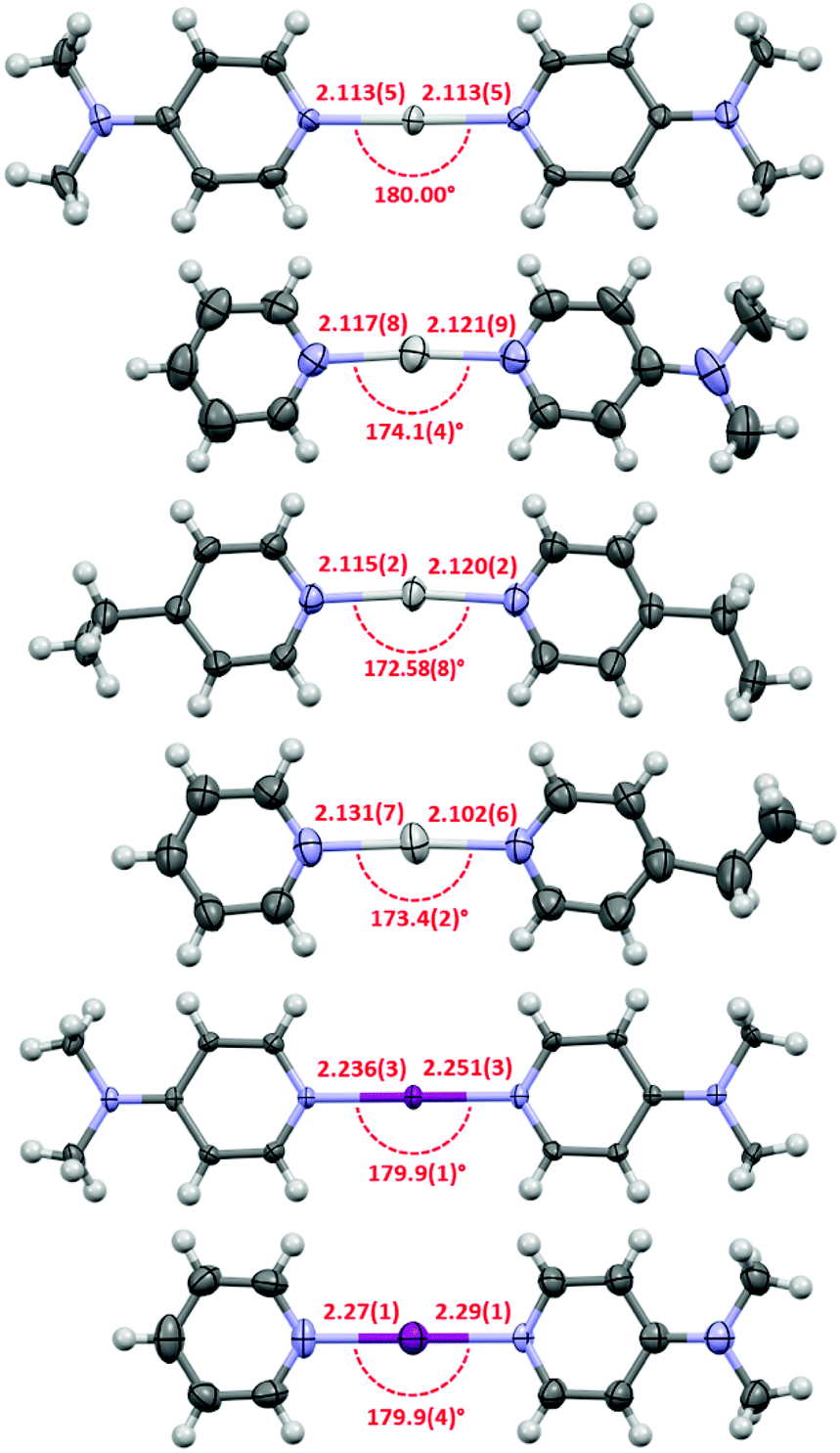 | ||
| Fig. 3 The X-ray crystal structures of 1b,§1c, 1d, 1e, 2b, and 2c (in order from top to bottom) with thermal displacement parameters at 50% probability (PF6 anions and solvates omitted for clarity; all lengths in Å). | ||
The symmetric silver complexes 1a,151b, and 1d exhibited Ag+–N bond lengths of 2.129, 2.077(7)–2.113(5),§ and 2.115(2)/2.120(2) Å, respectively, which were generally within experimental error of those observed for the asymmetric complexes 1c and 1e. A comparison of the pyridine I+–N bond lengths of 2c (2.29(1) Å) and 2a (2.268 Å)17 reveal that the bond length is only slightly elongated in the asymmetric complex, and likewise, comparison of the 4-DMAP I+–N bond lengths of 2c (2.27(1) Å) and 2b (2.236(3)/2.251(3) Å) once again showed a slight elongation in the asymmetric complex relative to its two symmetric analogues.
The packing of the complexes reveals that the silver complexes 1b, 1d, and 1e appear as dimers in the solid-state through weak argentophilic interactions (ESI†). Very surprisingly, and the first time in halonium complexes, also the iodonium complex 2c manifests shorter than vdW radii I+⋯I+ contacts (3.777 Å, ∑vdW = 3.96 Å), which is not unprecedented,26 though it has not been observed in unrestrained complexes such as [I(py)2][PF6].17
In order to probe this unusually short I+⋯I+ interaction of 2c that was observed in the solid state, it was studied computationally to interrogate its electronic origin. For this study DFT calculations combined with the quantum theory of atoms-in-molecules (QTAIM) analysis was used, as well as noncovalent interaction plots (NCI plot) and natural bond orbital (NBO) computational tools (see ESI† for computational methods and most of the results). To illustrate the argentophilic Ag+⋯Ag+ interaction observed in complexes 1b–e (see Fig. S34, ESI†) and compare it to the I+⋯I+ interaction of 2c, the NCI plot index has been used. The NCI plot is very convenient for the identification and visualisation of non-covalent interactions. This index is based on the peaks that appear in the reduced density gradient (RDG) at low densities. The formation of a supramolecular complex induces a significant change in the RDG at the critical points in between the interacting molecules due to the annihilation of the density gradient at these points.27,28 Consequently, the NCI plot is useful to evaluate host–guest complementarity and the extent to which noncovalent forces stabilise a complex. The information is basically qualitative showing which molecular regions interact. Red and blue colours are used to represent repulsive (ρcut+) and attractive (ρcut−) interactions, respectively. The yellow and green colours are used to represent weak repulsive and weak attractive forces, respectively. The representations of the NCI plots of dimers 1c (as representative example) and 2c are shown in Fig. 4. In both dimers, the NCI plot shows an extended and green isosurface that embraces both pyridine and dimethylaminopyridine ligands and also includes the region between both Ag or I atoms. In the dimer of 2c, the surface between both I+ is slightly better defined than the isosurface between the Ag+⋯Ag+ in 1c. It should be emphasised that the NCI plot index confirms the existence of the interactions and suggests that both the argentophilic and the I+⋯I+ interactions are attractive. The existence of these contacts has been also confirmed by QTAIM analysis by demonstrating the presence of a bond critical point and bond path connecting both Ag atoms in the dimers of compounds 1a–c (see Fig. S35, ESI†) and both I atoms in the dimer of 2c (see Fig. S36, ESI†).
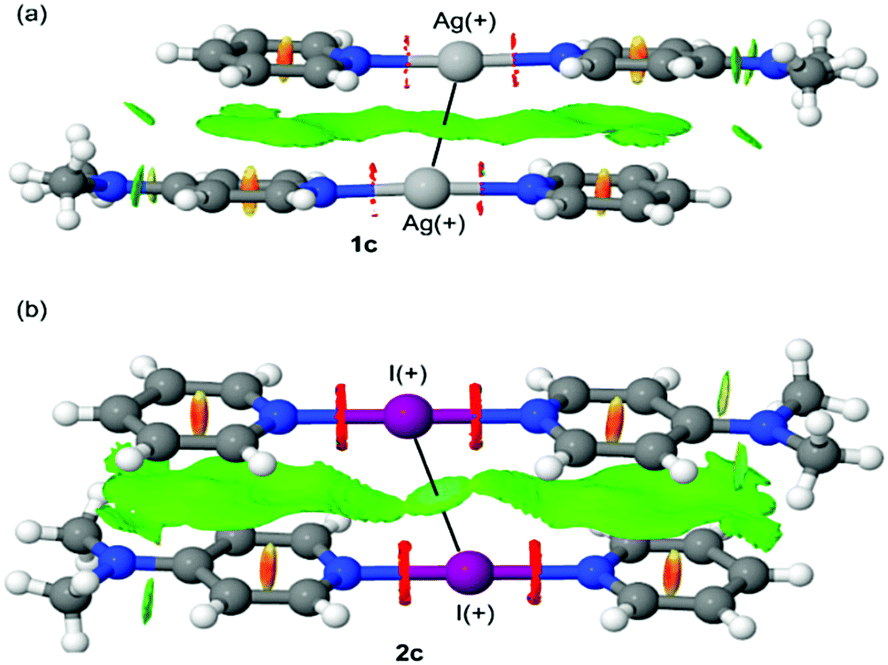 | ||
| Fig. 4 NCI plot of the dimers of 1c (a) and 2c (b). The gradient cut-off is s = 0.35 a.u., and the colour scale is −0.04 < ρ < 0.04 a.u. | ||
The asymmetry of the N–Ag/I distances in the compounds was also studied in the comprehensive theoretical study included in the ESI.† Unexpectedly, there are significant differences between the experimental and theoretical geometries (M06-2X/def2-TZVP level of theory). The comparison between the molecular orbitals between asymmetric Ag+ and I+ complexes revealed that there are significant differences between their HOMO and HOMO−1 plots (Fig. 5). That is, whilst the I+ atom actively participates in the MOs (via the pz AO conjugated with the π-systems of the arenes), the participation of the AOs of Ag+ is negligible. Moreover, the HOMO and HOMO−1 in 2c (Fig. 5b) reveal π-antibonding character in the I+–N bond, thus explaining the longer I–N distances compared to Ag–N ones. This is also noticed in the NCI plot (Fig. 4b) that shows a red belt-shaped surface around the σ-bond. The DFT calculations suggest that the asymmetry of the Ag/I–N distances observed in the X-ray structures of Fig. 3 and their differences with the theoretical ones (see Fig. S40 and S41, ESI†) are likely due to packing effects.
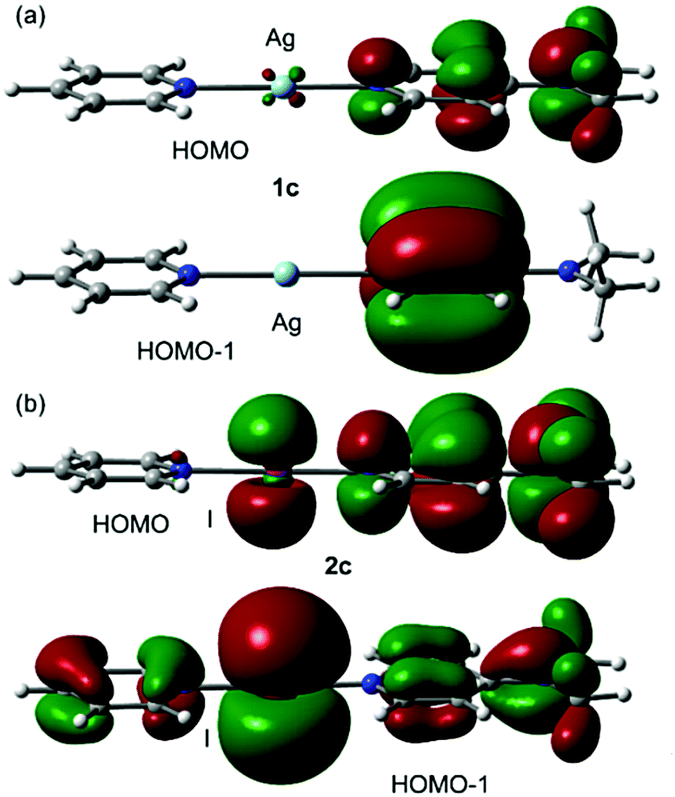 | ||
| Fig. 5 HOMO and HOMO−1 plots of compounds 1c (a) and 2c (b). | ||
In conclusion, the first asymmetric linear silver complexes, [Ag(py)(4-DMAP)]PF6 (1c) and [Ag(py)(4-Etpy)]PF6 (1e), and the first asymmetric halonium complexes, [I(py)(4-DMAP)]PF6 (2c) and [I(py)(4-Etpy)]PF6 (2e), were prepared and their identity confirmed by 1H and 1H–15N HMBC NMR spectroscopy, and for 1c, 1e, and 2c, further confirmed in the solid-state by X-ray crystallography. The argentophilic Ag+⋯Ag+ interactions of 1c and 1e, and close I+⋯I+ contact of 2c have been computationally interrogated and analysed. These novel asymmetric complexes were further analysed by comparison to their respective homoleptic counterparts, which demonstrated that the asymmetric complexes persist in the solution state, and can be observed by 1H and correlated 1H–15N NMR spectroscopy.
Financial support by MICIU/AEI of Spain (AF, project number CTQ2017-85821-R, FEDER funds) and The Finnish Cultural Foundation Central Fund (JSW, grant number 00201148) is gratefully acknowledged.
Conflicts of interest
There are no conflicts to declare.Notes and references
- G. Cavallo, P. Metrangolo, R. Milani, T. Pilati, A. Priimagi, G. Resnati and G. Terraneo, Chem. Rev., 2016, 116, 2478–2601 CrossRef CAS PubMed.
- L. C. Gilday, S. W. Robinson, T. A. Barendt, M. J. Langton, B. R. Mullaney and P. D. Beer, Chem. Rev., 2015, 115, 7118–7195 CrossRef CAS PubMed.
- G. R. Desiraju, P. S. Ho, L. Kloo, A. C. Legon, R. Marquardt, P. Metrangolo, P. Politzer, G. Resnati and K. Rissanen, Pure Appl. Chem., 2013, 85, 1711–1713 CAS.
- K. Rissanen, CrystEngComm, 2008, 10, 1107–1113 RSC.
- L. Turunen and M. Erdélyi, Chem. Soc. Rev., 2020, 49, 2688–2700 RSC.
- J. A. Creighton, I. Haque and J. L. Wood, Chem. Commun., 1966, 229 RSC.
- I. Haque and J. L. Wood, J. Mol. Struct., 1968, 2, 217–238 CrossRef CAS.
- J. Barluenga, J. M. González, M. A. Garcia-Martin, P. J. Campos and G. Asensio, J. Chem. Soc., Chem. Commun., 1992, 1016–1017 RSC.
- J. Ezquerra, A. C. Pedregal, C. Lamas, J. Barluenga, M. Pérez, A. M. A. García-Martín and J. M. González, J. Org. Chem., 1996, 61, 5804–5812 CrossRef CAS.
- G. Espuña, G. Arsequell, G. Valencia, J. Barluenga, M. Pérez and J. M. González, Chem. Commun., 2000, 1307–1308 RSC.
- A. Yoshimura and V. V. Zhdankin, Chem. Rev., 2016, 116, 3328–3435 CrossRef CAS PubMed.
- S. Ghosh, S. Pradhan and I. Chatterjee, Beilstein J. Org. Chem., 2018, 14, 1244–1262 CrossRef CAS PubMed.
- J. C. Dyason, P. C. Healy, L. M. Engelhardt and A. H. White, Aust. J. Chem., 1985, 38, 1325–1328 CrossRef CAS.
- S. Gotsis and A. H. White, Aust. J. Chem., 1987, 40, 1603–1608 CrossRef CAS.
- C. Y. Chen, J. Y. Zeng and H. M. Lee, Inorg. Chim. Acta, 2007, 360, 21–30 CrossRef CAS.
- Y. Kim, E. J. Mckinley, K. E. Christensen, N. H. Rees and A. L. Thompson, Cryst. Growth Des., 2014, 14, 6294–6301 CrossRef CAS.
- M. Bedin, A. Karim, M. Reitti, A.-C. C. Carlsson, F. Topić, M. Cetina, F. Pan, V. Havel, F. Al-Ameri, V. Sindelar, K. Rissanen, J. Gräfenstein and M. Erdélyi, Chem. Sci., 2015, 6, 3746–3756 RSC.
- S. Lindblad, K. Mehmeti, A. X. Veiga, B. Nekoueishahraki, J. Gräfenstein and M. Erdélyi, J. Am. Chem. Soc., 2018, 140, 13503–13513 CrossRef CAS PubMed.
- A.-C. C. Carlsson, K. Mehmeti, M. Uhrbom, A. Karim, M. Bedin, R. Puttreddy, R. Kleinmaier, A. A. Neverov, B. Nekoueishahraki, J. Gräfenstein, K. Rissanen and M. Erdélyi, J. Am. Chem. Soc., 2016, 138, 9853–9863 CrossRef CAS PubMed.
- K. Rissanen and M. Haukka, Top. Curr. Chem., 2015, 359, 77–90 CrossRef CAS PubMed.
- L. Koskinen, P. Hirva, E. Kalenius, S. Jääskeläinen, K. Rissanen and M. Haukka, CrystEngComm, 2015, 17, 1231–1236 RSC.
- L. Turunen, U. Warzok, R. Puttreddy, N. K. Beyeh, C. A. Schalley and K. Rissanen, Angew. Chem., Int. Ed., 2016, 55, 14033–14036 CrossRef CAS PubMed.
- L. Turunen, A. Peuronen, S. Forsblom, E. Kalenius, M. Lahtinen and K. Rissanen, Chem. – Eur. J., 2017, 23, 11714–11718 CrossRef CAS PubMed.
- L. Turunen, U. Warzok, C. A. Schalley and K. Rissanen, Chem, 2017, 3, 861–869 CAS.
- U. Warzok, M. Marianski, W. Hoffmann, L. Turunen, K. Rissanen, K. Pagel and C. A. Schalley, Chem. Sci., 2018, 9, 8343–8351 RSC.
- A. Vanderkooy, A. K. Gupta, T. Földes, S. Lindblad, A. Orthaber, I. Pápai and M. Erdélyi, Angew. Chem., Int. Ed., 2019, 58, 9012–9016 CrossRef CAS PubMed.
- J. Contreras-García, E. R. Johnson, S. Keinan, R. Chaudret, J.-P. Piquemal, D. N. Beratan and W. Yang, J. Chem. Theory Comput., 2011, 7, 625–632 CrossRef PubMed.
- E. R. Johnson, S. Keinan, P. Mori-Sánchez, J. Contreras-García, A. J. Cohen and W. Yang, J. Am. Chem. Soc., 2010, 132, 6498–6506 CrossRef CAS PubMed.
- D. C. Georgiou, P. Butler, E. C. Browne, D. J. D. Wilson and J. L. Dutton, Aust. J. Chem., 2013, 66, 1179–1188 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Synthesis and NMR details. CCDC 1996937–1996944. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0cc02758h |
| ‡ [I(4-DMAP)2][NO3] has been previously reported.29 |
| § Three different X-ray structures were obtained for 1b. |
| This journal is © The Royal Society of Chemistry 2020 |