
Efficient near-infrared luminescence from bis-cyclometalated iridium(III) complexes with rigid quinoline-derived ancillary ligands†
Po-Ni
Lai
 ,
Sungwon
Yoon
and
Thomas S.
Teets
*
,
Sungwon
Yoon
and
Thomas S.
Teets
*
Department of Chemistry, University of Houston, 3585 Cullen Blvd. Room 112, Houston, TX 77204-5003, USA. E-mail: tteets@uh.edu
First published on 12th June 2020
Abstract
Five new near-infrared (NIR) phosphorescent bis-cyclometalated iridium(III) complexes, partnering highly conjugated cyclometalating ligands with quinoline-derived ancillary ligands, have been developed. These complexes have peak NIR luminescence wavelengths from 711 to 729 nm, with photoluminescence quantum yields ranging from 0.042 to 0.36.
Phosphorescent cyclometalated iridium(III) complexes are among the most widely explored class of phosphorescent materials with applications in everything from sensing,1 to photoredox catalysis,2 to organic light-emitting diodes (OLEDs),3 and light-emitting electrochemical cells (LEECs).4 Within this family of materials, heteroleptic complexes of the Ir(C∧N)2(L∧X) structure type (C∧N = cyclometalating ligand, L∧X = ancillary ligand) are very well-developed due to their ease of synthesis and their high photoluminescence efficiencies, tunable emission wavelengths, and high thermal and photostability.5,6
In the past few decades, most research has focused on the development of new cyclometalated iridium complexes to cover the whole visible spectrum, given the widespread and lucrative applications of these compounds in color displays. Beyond the visible spectrum into the near-infrared (NIR) (700–900 nm), iridium(III) complexes with high photoluminescence quantum yields are relatively rare.7 NIR-emitting materials have aroused particular interest in electroluminescent diodes promising for military optoelectronics and telecommunications,8 as well as emerging applications in food safety.9 In addition, this spectral window is particularly desirable for biological applications because of decreased light scattering, minimal background absorption and photodamage, and improved tissue penetration depth.10,11 While most of the available NIR probes are organic dyes,12 quantum dots (QDs),13 metal macrocyclic complexes,14 and lanthanide-based phosphors,15,16 it is clear that there is a demand for further development of novel NIR luminescent materials for these applications.
The design of near-infrared phosphorescent materials that exhibit high photoluminescence quantum yields (ΦPL) is intrinsically more difficult owing to their dependence on the energy gap law. According to the energy gap law, the inverse of the nonradiative rate constant, knr, has a natural exponential dependence on the excited-state energy,17 meaning knr increases as the luminescence shifts into the NIR region. Also, from second-order perturbation theory, the cubic dependence of radiative rate constant (kr) on the excited-state energy makes kr smaller for lower-energy emitters, which likewise contributes to the typically low quantum yields.3 NIR luminescence in cyclometalated iridium complexes is typically accomplished by using highly conjugated cyclometalating ligands to lower the triplet state (T1) energy, most often in bis-cyclometalated structures with acetylacetonate (acac) or bipyridine derivatives as ancillary ligands.18–20 We have previously shown in red and deep-red phosphorescent complexes that electron-rich, π-donating ancillary ligands strongly perturb the metal-centered HOMO, increase the metal d-orbital participation in the excited state, and as a result augment kr and ΦPL.21–23 With this strategy, we have achieved ΦPL values as high as 0.8 in the red region of the spectrum.21,22
A similar strategy should be effective in the near-infrared region, and herein we report five new neutral near-infrared phosphorescent iridium complexes with a phenanthridine-benzothiophene (btph) cyclometalating ligand (C∧N), previously used to support complexes applied as imaging probes for cancer cells.24–27 In our previous work in the red and deep red regions, we used ancillary ligands primarily from the β-ketoiminate, β-diketiminate, amidinate, and amidate families, which had beneficial effects on kr but in general gave little control over knr. Furthermore, these ancillary ligands were generally not effective when the cyclometalated aryl ring is a thiophene derivative, as is the case with btph. In this work we opted to combine strong donor moieties on the ancillary ligand with a rigid fused aromatic skeleton, which could potentially suppress knr and thus enhance the function of NIR phosphors. We reasoned that 8-substituted quinolines and 10-substituted benzoquinolines, previously used on rare occasions in the design of phosphorescent iridium complexes,28–30 could be good supporting ligands for NIR emitters for these reasons.
The syntheses of five new iridium(III) complexes and their chemical structures are illustrated in Scheme 1, together with the abbreviations used in this communication. The C∧N ligand, btphH, and the μ-chloro-bridge iridium dimer precursor [Ir(btph)2(μ-Cl)2]2 were synthesized by known procedures.31 Three distinct but related methods were used to install the ancillary ligand. Complexes with 8-hydroxyquinoline (8OQ, 1), 8-carboxyquinoline (8COOQ, 2) and 10-hydroxybenzo[h]quinoline (10OBQ, 3) were synthesized using dichloromethane and ethanol mixture as the solvent and triethylamine as the base. Complex 4, containing 8-(1H-pyrrol-2-yl)quinoline (8PyQ) was made using NaH as the base and THF as the solvent. Complex 5, with N-phenyl-8-quinolinecarboxamide (8CONPhQ) as the ancillary ligand, was synthesized by combining the potassium salt of 8CONPhQ with [Ir(btph)2(μ-Cl)2]2. The Ir(btph)2(L∧X) products were isolated in 12–61% yield. All complexes were characterized by 1H NMR (Fig. S8–S12 in the ESI†), but their poor solubility precludes 13C{1H} NMR analysis.
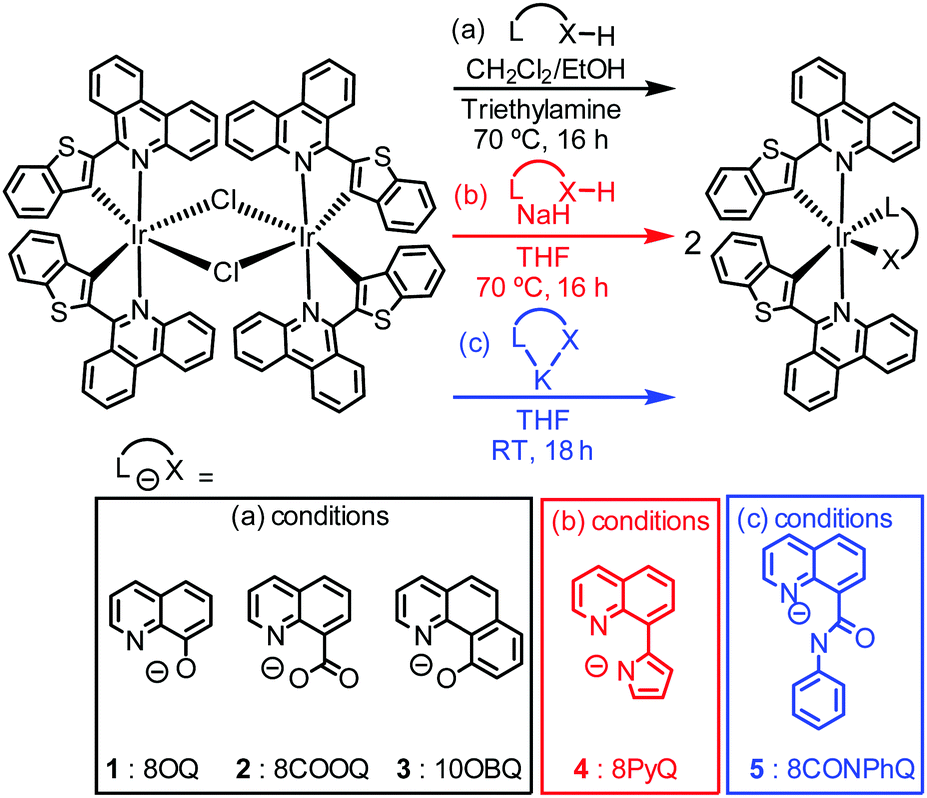 | ||
| Scheme 1 Synthesis of iridium complexes described here. | ||
Single crystals of complexes 1 and 2 suitable for single-crystal X-ray diffraction measurements were grown via the slow diffusion of hexane vapor into a dichloromethane solution of the complex, and their structures are shown in Fig. 1 with refinement data summarized in Table S1 of the ESI.† The complexes feature a distorted octahedral iridium(III) center with phenanthridine moieties from both cyclometalating ligands in a trans orientation. The btph ligands are nonplanar, with a slight twist along the C–C bond connecting the benzothiophene and phenanthridine to avoid interligand steric clash of the latter with the quinoline ancillary ligand, and a slight buckling of the phenanthridine to avoid intraligand steric hindrance. The ancillary ligand bite angles ∠N(3)–Ir(1)–O(1) for the complexes 1 and 2 are 77.32(14)° and 82.00(10)°, respectively, similar to the typical five- and six-membered chelate complexes reported by our group.22
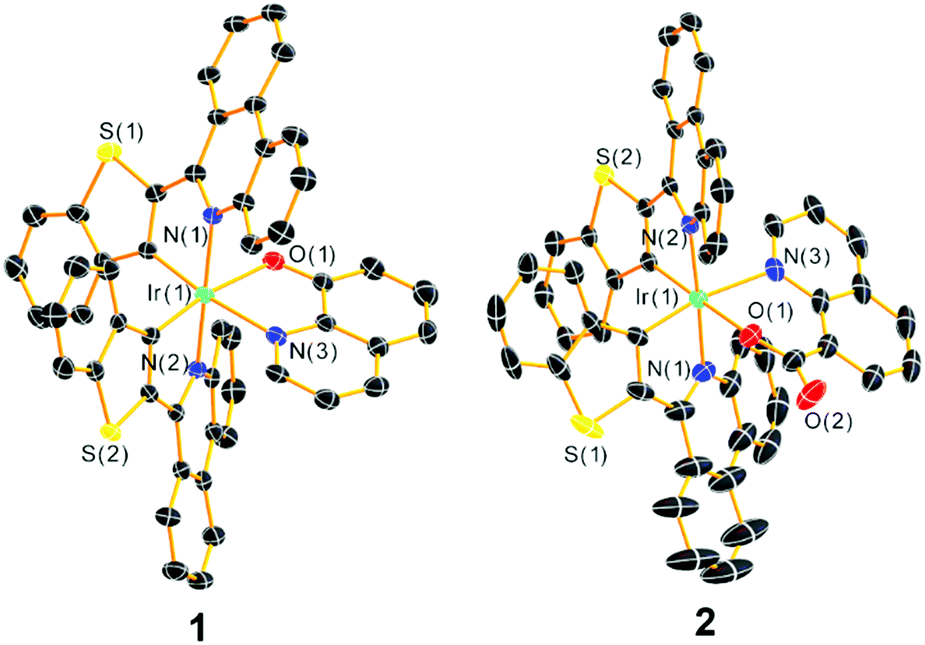 | ||
| Fig. 1 Molecular structures of 1 and 2 determined from single-crystal X-ray diffraction. Thermal ellipsoids are drawn at the 50% probability level with solvent molecules and hydrogen atoms eliminated. | ||
The UV-vis absorption spectra are shown in Fig. 2, and provide some insight into the electronic structure and frontier orbitals. In the UV-vis absorption spectra of complexes 1–5, there are three distinct spectral regions. Intense bands from 290 nm to 350 nm with large molar extinction coefficients (ε = 16–86 × 103 M−1 cm−1) were observed, assigned to spin-allowed, localized ligand-centered (LC) π–π* transition of the btph cyclometalating ligands and quinoline ancillary ligands. Overlapping absorption bands in the near-UV and blue regions, ca. 350–475 nm, correspond to delocalized 1LC transitions. Finally, the visible absorption bands, centered around 520–568 nm and tailing beyond 600 nm are assigned to metal-to-ligand charge transfer (1MLCT) states, originating from a dπ(Ir)–π*(btph) HOMO → LUMO transition Complex 4 exhibits an additional and rather weak longer wavelength absorption, at ca. 680 nm, which is assigned to a spin-forbidden transition to a 3MLCT excited state, i.e. an S0 → T1 transition. Further support for the latter assignments come from cyclic voltammetry studies, summarized in Table 1 and shown in Fig. S1 and S2 of the ESI.† CVs of most complexes, except 5, show two reduction waves with potentials of ca. −2.1 and −2.4 V vs. Fc+/Fc, which involve successive population of btph π* orbitals that are very similar in energy across the series. In contrast, the formally IrIV/IrIII oxidation potentials, at similar values of +0.34 to +0.46 V for complexes 1, 2, 3, and 5, shifts dramatically to +0.05 V in complex 4, where the ancillary ligand includes a more basic pyrrolide donor, suggesting the HOMO energy is sensitive to the donor characteristics of the ancillary ligand. Nevertheless, the HOMO–LUMO gaps for the five complexes do not vary dramatically, and as a result their visible 1MLCT transitions are all similar and don’t seem to shift systematically with the ancillary ligand.
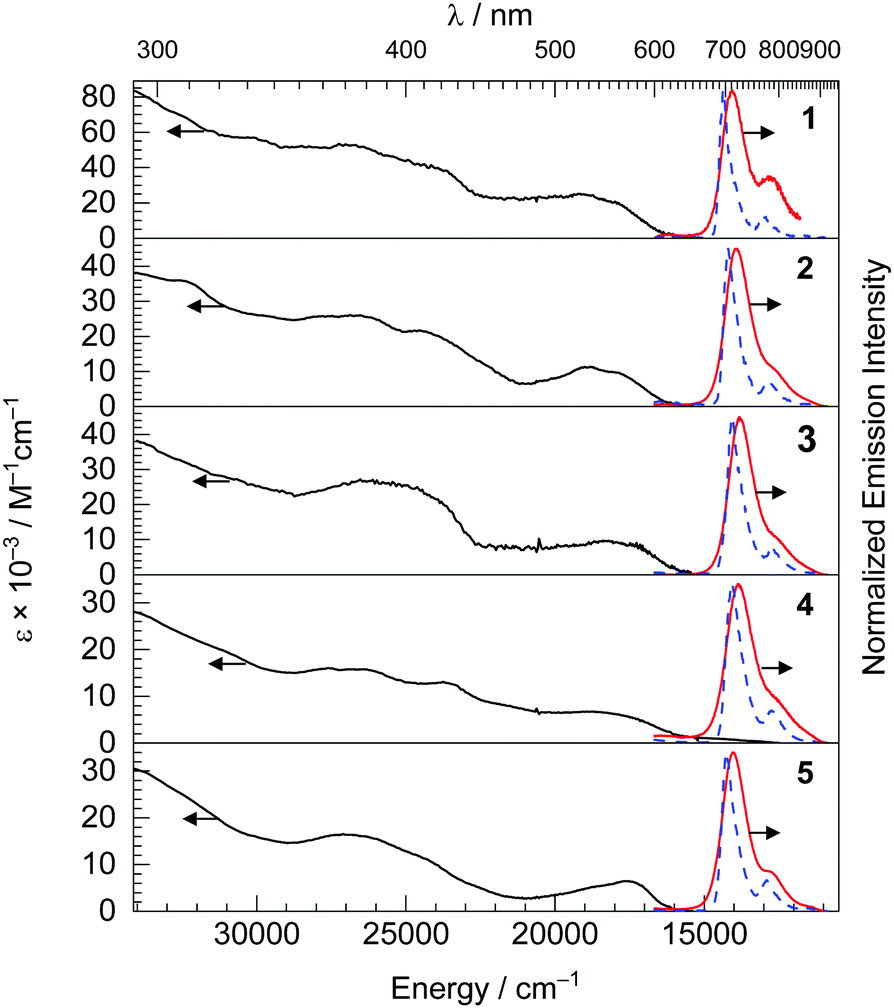 | ||
| Fig. 2 Stacked plots of UV-vis absorption and room temperature PL emission spectra were measured in deaerated THF (black and red solid lines), along with 77 K PL emission spectra measured in toluene glass (blue dashed lines). | ||
| λ max (nm) | Φ PL | τ (μs) | k r × 10−5 (s−1)/knrc × 10−5 (s−1) | FWHMd (cm−1) 293 K/77 K | S M | ħω M (cm−1) | E(IrIV/IrIII) (V) | E red (V) | ||
|---|---|---|---|---|---|---|---|---|---|---|
| THF at 293 Ka | Toluene at 77 Ka | |||||||||
| a λ ex = 500 nm. b λ ex = 430 nm. c k r = ΦPL/τ and knr = (1 − ΦPL)/τ. d The FWHM for the first vibronic band was determined at both 293 K and 77 K, and the Huang–Rhys factor, SM, was estimated from the peak heights of the two vibronic bands in the 77 K PL data. e The energy of ħωM was obtained from the energy difference of the first two emission peaks. f Irreversible features are quoted as Ep,a or Ep,c values. | ||||||||||
| 1 | 711 | 695 | 0.36 | 2.0 | 1.8/3.2 | 960/410 | 0.14 | 1340 | +0.40f | −2.08, −2.36 |
| 2 | 724 | 704 | 0.30 | 1.7 | 1.8/4.1 | 1030/520 | 0.15 | 1360 | +0.46f | −2.04, −2.25f |
| 3 | 723 | 711 | 0.28 | 1.5 | 1.9/4.8 | 1050/550 | 0.15 | 1390 | +0.34 | −2.04, −2.38 |
| 4 | 729 | 711 | 0.042 | 1.0 | 0.42/9.6 | 1110/620 | 0.19 | 1300 | +0.05f | −2.10, −2.39 |
| 5 | 716 | 702 | 0.34 | 1.6 | 2.1/4.1 | 1060/520 | 0.18 | 1330 | +0.42 | −2.16 |
Steady-state room-temperature emission spectra of the five iridium(III) complexes were recorded in deoxygenated THF with 500 nm excitation, and they are displayed in Fig. 2 with major spectroscopic characteristics summarized in Table 1. The excitation spectra of the new complexes were also collected and shown in the Fig. S3–S7 (ESI†), and in each case are superimposed with the absorption spectrum indicating that luminescence arises solely from the iridium complex. The emission maxima of all the complexes are in the NIR region, spanning 711–729 nm with the pyrrole complex 4 having the largest red shift of 11 nm and hydroxyl complex 1 the largest blue-shift of 7 nm compared to the peak emission wavelength of Ir(btph)2(acac) (λmax = 718 nm).31
At room temperature, the photoluminescence quantum yield ΦPL of most of the complexes (0.28–0.36) are similar to or slightly higher than the reference complex Ir(btph)2(acac) (ΦPL = 0.28),31 except for complex 4 where ΦPL = 0.042, which shows that the rigid quinoline-based ancillary ligands can support efficient NIR luminescence. The PL decays are all single-exponential with lifetimes ranging from 1.0 to 2.0 μs. To understand the lower ΦPL in complex 4, we note that its kr is more than four-fold smaller and its knr is more than two-fold larger than the other members of the series. For the remaining compounds with high quantum yields, the knr values reported here are smaller than those of most other structurally related deep-red and NIR phosphorescent compounds,19,23,32 suggesting the rigidity of the btph C∧N ligands and quinoline-based ancillary ligands combine to inhibit vibrational relaxation pathways. The closely related reference compound Ir(btph)2(acac) has a quantum yield of 0.28 and a lifetime of 1.9 μs.31 In comparison, the most efficient emitter in this series is complex 1, where ΦPL = 0.36. The increase in quantum yield is a result of a slightly larger kr (1.8 vs. 1.5 × 105 s−1) and slightly smaller knr (3.2 vs. 3.8 × 105 s−1) in complex 1. One consistent trend is that in all of 1, 2, 3, and 5, where the quantum yields are equal to or slightly larger than Ir(btph)2(acac), we observe slightly larger kr values, likely an effect of the more electron-rich donors in the ancillary ligands of most of these compounds.21,22 That said, in most members of the series the PL dynamics are quite similar to one another and to Ir(btph)2(acac), indicating that the ancillary ligand does not generally have a large influence on the radiative and nonradiative decay from the triplet state.
The emission spectra were also recorded in a frozen toluene glass at 77 K. Comparing to the room-temperature spectra, small rigidochromic shifts (230–390 cm−1) are observed. In the emission spectra at 77 K, two distinct vibronic maxima can be clearly resolved. As shown in Table 1, we determined the full-width at half-maximum (FWHM) values of the high energy vibronic band at both 293 K and 77 K. The FWHMs are rather narrow, spanning 960–1110 cm−1 at 293 K, and are approximately halved at 77 K, ranging from 410–620 cm−1. From the energy difference between the E0–0 and E0–1 peaks, the ground-state vibrational quantum spacing (ħωM value) was also calculated, ranging from 1300 to 1390 cm−1 and consistent with aromatic stretching vibrations in the btph ligand. Taken together, these observations from the 77 K PL spectra indicate that the emissive T1 state is predominantly 3LC in nature. The Huang–Rhys factor (SM), used to evaluate the degree of structural distortion that occurs in the excited state relative to the ground state, can also be estimated from the 77 K PL data. The SM values are 0.14–0.19, indicating weak coupling between the excited and ground-state vibrational modes and consistent with the notion that the rigid ligand structures and extensive conjugation in these compounds result in a minimally distorted triplet state.
In summary, five novel bis-cyclometalated iridium near-infrared phosphorescent emitters have been synthesized and characterized. Four of the complexes have intense NIR emission with photoluminescence quantum yields that rival or exceed state-of-the-art NIR phosphors. We demonstrate that the rigid btph cyclometalating ligands, in combination with substituted quinoline ancillary ligands, result in narrow emission bands in the NIR with excited-state dynamics favorable for efficient luminescence. The compounds we introduce are promising candidates for further applications in night-vision technology, medical imaging, and biological probes and sensors.
The authors acknowledge the National Science Foundation (grant no. CHE-1846831) and the Welch Foundation (grant no. E-1887) for funding this research. The authors thank Dr.Po-An Chen for helpful discussions regarding the syntheses.
Conflicts of interest
There are no conflicts to declare.Notes and references
- X. Wang and O. S. Wolfbeis, Chem. Soc. Rev., 2014, 43, 3666–3761 RSC.
- C. K. Prier, D. A. Rankic and D. W. C. MacMillan, Chem. Rev., 2013, 113, 5322–5363 CrossRef CAS PubMed.
- H. Yersin, Highly Efficient OLEDs with Phosphorescent Materials, John Wiley & Sons, 2008 Search PubMed.
- R. D. Costa, E. Ortí, H. J. Bolink, F. Monti, G. Accorsi and N. Armaroli, Angew. Chem., 2012, 124, 8300–8334 CrossRef.
- Q. Zhao, F. Li and C. Huang, Chem. Soc. Rev., 2010, 39, 3007–3030 RSC.
- Q. Zhao, C. Huang and F. Li, Chem. Soc. Rev., 2011, 40, 2508–2524 RSC.
- T.-R. Chen, J. Organomet. Chem., 2008, 693, 3117–3130 CrossRef CAS.
- J. Clark and G. Lanzani, Nat. Photonics, 2010, 4, 438–446 CrossRef CAS.
- X. Fu and Y. Ying, Crit. Rev. Food Sci. Nutr., 2016, 56, 1913–1924 CrossRef CAS PubMed.
- Near-infrared applications in biotechnology, ed. R. Raghavachari and M. Dekker, New York, 2001 Search PubMed.
- Z. Guo, S. Park, J. Yoon and I. Shin, Chem. Soc. Rev., 2014, 43, 16–29 RSC.
- X. Zhao, Y. Li, D. Jin, Y. Xing, X. Yan and L. Chen, Chem. Commun., 2015, 51, 11721–11724 RSC.
- F. D. Duman, I. Hocaoglu, D. G. Ozturk, D. Gozuacik, A. Kiraz and H. Yagci Acar, Nanoscale, 2015, 7, 11352–11362 RSC.
- Y. Yao, C.-L. Hou, Z.-S. Yang, G. Ran, L. Kang, C. Li, W. Zhang, J. Zhang and J.-L. Zhang, Chem. Sci., 2019, 10, 10170–10178 RSC.
- M. Lin, Y. Zhao, S. Wang, M. Liu, Z. Duan, Y. Chen, F. Li, F. Xu and T. Lu, Biotechnol. Adv., 2012, 30, 1551–1561 CrossRef CAS PubMed.
- J.-Y. Hu, Y. Ning, Y.-S. Meng, J. Zhang, Z.-Y. Wu, S. Gao and J.-L. Zhang, Chem. Sci., 2017, 8, 2702–2709 RSC.
- R. Englman and J. Jortner, Mol. Phys., 1970, 18, 145–164 CrossRef CAS.
- R. Tao, J. Qiao, G. Zhang, L. Duan, L. Wang and Y. Qiu, J. Phys. Chem. C, 2012, 116, 11658–11664 CrossRef CAS.
- C.-L. Ho, H. Li and W.-Y. Wong, J. Organomet. Chem., 2014, 751, 261–285 CrossRef CAS.
- Z. Chen, H. Zhang, D. Wen, W. Wu, Q. Zeng, S. Chen and W.-Y. Wong, Chem. Sci., 2020, 11, 2342–2349 RSC.
- P.-N. Lai, C. H. Brysacz, M. K. Alam, N. A. Ayoub, T. G. Gray, J. Bao and T. S. Teets, J. Am. Chem. Soc., 2018, 140, 10198–10207 CrossRef CAS PubMed.
- P. Lai and T. S. Teets, Chem. – Eur. J., 2019, 25, 6026–6037 CrossRef CAS PubMed.
- E. Kabir, Y. Wu, S. Sittel, B.-L. Nguyen and T. S. Teets, Inorg. Chem. Front., 2020, 7, 1362–1373 RSC.
- S. Zhang, M. Hosaka, T. Yoshihara, K. Negishi, Y. Iida, S. Tobita and T. Takeuchi, Cancer Res., 2010, 70, 4490–4498 CrossRef CAS PubMed.
- T. Yoshihara, S. Murayama, T. Masuda, T. Kikuchi, K. Yoshida, M. Hosaka and S. Tobita, J. Photochem. Photobiol., A, 2015, 299, 172–182 CrossRef CAS.
- X. Zheng, X. Wang, H. Mao, W. Wu, B. Liu and X. Jiang, Nat. Commun., 2015, 6, 5834 CrossRef CAS PubMed.
- I. S. Kritchenkov, P. S. Chelushkin, V. V. Sokolov, V. V. Pavlovskiy, V. V. Porsev, R. A. Evarestov and S. P. Tunik, Organometallics, 2019, 38, 3740–3751 CrossRef CAS.
- J. Guo, J. Zhou, G. Fu, Y. He, W. Li and X. Lü, Inorg. Chem. Commun., 2019, 101, 69–73 CrossRef CAS.
- S. Kappaun, S. Sax, S. Eder, K. C. Möller, K. Waich, F. Niedermair, R. Saf, K. Mereiter, J. Jacob, K. Müllen, E. J. W. List and C. Slugovc, Chem. Mater., 2007, 19, 1209–1211 CrossRef CAS.
- H.-R. Park and Y. Ha, J. Nanosci. Nanotechnol., 2012, 12, 1365–1370 CrossRef CAS PubMed.
- T. Yoshihara, S. Murayama, T. Masuda, T. Kikuchi, K. Yoshida, M. Hosaka and S. Tobita, J. Photochem. Photobiol., A, 2015, 299, 172–182 CrossRef CAS.
- G. Zhang, H. Zhang, Y. Gao, R. Tao, L. Xin, J. Yi, F. Li, W. Liu and J. Qiao, Organometallics, 2014, 33, 61–68 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details, X-ray crystallography summary tables, cyclic voltammograms, excitation spectra, and NMR spectra. CCDC 1995456 (1) and 1995457 (2) contain the crystallographic data. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0cc02745f |
| This journal is © The Royal Society of Chemistry 2020 |