DOI:
10.1039/D0CC02660C
(Feature Article)
Chem. Commun., 2020,
56, 9256-9267
Formaldehyde tert-butyl hydrazone as a formyl anion equivalent: asymmetric addition to carbonyl compounds†
Received
13th April 2020
, Accepted 23rd June 2020
First published on 23rd June 2020
Abstract
The asymmetric 1,2-addition of formyl anion equivalents to carbonyl compounds is a powerful synthetic tool that ideally provide access to highly functionalizable α-hydroxy aldehydes in an enantioselective fashion. In this context, the nucleophilic character of formaldehyde hydrazones, together with their remarkable stability as monomeric species, has been exploited for the functionalization of diverse carbonyl compounds, using initially auxiliary-based methodologies and, more recently, catalytic enantioselective versions. This feature article highlights our research progress employing formaldehyde tert-butyl hydrazone as a versatile formyl anion equivalent, in combination with bifunctional H-bonding organocatalysis. The design and optimization of different catalytic systems, focusing on a dual activation of both reagents, is reviewed, as well as the racemization free unmasking of the formyl group and representative product transformations for the construction of valuable, densely functionalyzed chiral building blocks.

Esteban Matador
| Esteban Matador studied chemistry at the University of Sevilla where he received his BSc degree (2014) and MSc degree (2015). He is currently performing PhD studies in the research group of Prof. Rosario Fernández and José M. Lassaletta. During this period, he spent pre-doc stages in the group of Prof. Magnus Rueping at the Institut of Organic Chemistry at the RWTH Aachen University (Germany, 2017), and in the group of Prof. Sander J. Wezenberg at the Leiden Institute of Chemistry of the Leiden University (The Netherlands, 2020). His research interests include asymmetric homogeneous catalysis, and development of new synthetic methodologies, with emphasis on hydrazones as reagents in organocatalysis. |

María de Gracia Retamosa
| María de Gracia Retamosa received her PhD in 2008 at the University of Alicante (Spain) under the guidance of Prof. Carmen Nájera and José Miguel Sansano. After that she undertook several postdoctoral positions [Prof. Michael Greaney at University of Edimburgh (UK, 2009), Prof. Jesús M. Sanz at University Miguel Hernández (Elche, Spain, 2009–2011) and Prof. Fernando P. Cossío at the University of the Basque Country and Donostia International Physics Center (Spain, 2012–2016)]. In February 2016 she joined the group of Prof. Rosario Fernández and José M. Lassaletta [IIQ-CSIC (Sevilla, Spain)], and was promoted to Associate researcher in 2019 at the University of Seville. Her current research interests include asymmetric metal catalysis, organocatalysis and synthesis of compounds with pharmacological interest. |

David Monge
| David Monge received his PhD in 2007 at the University of Sevilla under the supervision of Prof. Rosario Fernández and José M. Lassaletta. After that he was postdoctoral researcher at CSIC (Sevilla) for BayerCropScience GmBH (2007–2008) and he joined the group of Prof. Karl Anker Jørgensen at Center for Catalysis at the University of Aarhus (Denmark, 2009–2010). In 2011 he returned to University of Sevilla, where he was a postdoctoral “Juan de la Cierva” fellow, and was promoted to Associate Researcher in 2015 and Associate Professor in 2019. His current research interests include green chemistry and asymmetric metal catalysis and organocatalysis, with emphasis on hydrazones as reagents or ligands. |

Rosario Fernández
| Rosario Fernández studied chemistry at the University of Sevilla and received both her BS degree (1980) and her PhD degree (1985) under the supervision of Prof. Antonio Gómez Sánchez. She was a NATO postdoctoral fellow at the University of Paris-Sud (Orsay, France) in the laboratory of Prof. Serge David from 1986 to 1987. In 1987 she returned to the University of Sevilla, where she was promoted to Associate Professor. In 2008 she became a Full Professor at the same University. Her current research interests include asymmetric synthesis and enantioselective catalysis, in both aspects, asymmetric metal catalysis and organocatalysis. |

José M. Lassaletta
| José María Lassaletta received his PhD in 1990 under the supervision of Prof. Gómez-Guillén at the University of Seville. After a postdoctoral stage in the ‘Instituto de la Grasa y sus Derivados’ (CSIC) he joined the group of Professor Richard R. Schmidt (U. Konstanz, Germany). In 1995 he moved to the Instituto de Investigaciones Químicas (CSIC, Seville), where he promoted to Tenured Scientist in 1996, Research Scientist in 2005 and Research Professor in 2009. He has been recognized with the ‘Felix Serratosa’ Lecture (2011) and the ‘Ignacio Ribas’ Medal from the Organic Chemistry Division of the Royal Society of Chemistry (2017). He is currently interested in the development of synthetic methodologies, cross-coupling & C–H activation strategies, ligand design, with emphasis in hydrazones & N-heterocyclic carbenes, and asymmetric organocatalysis. |
1. Introduction
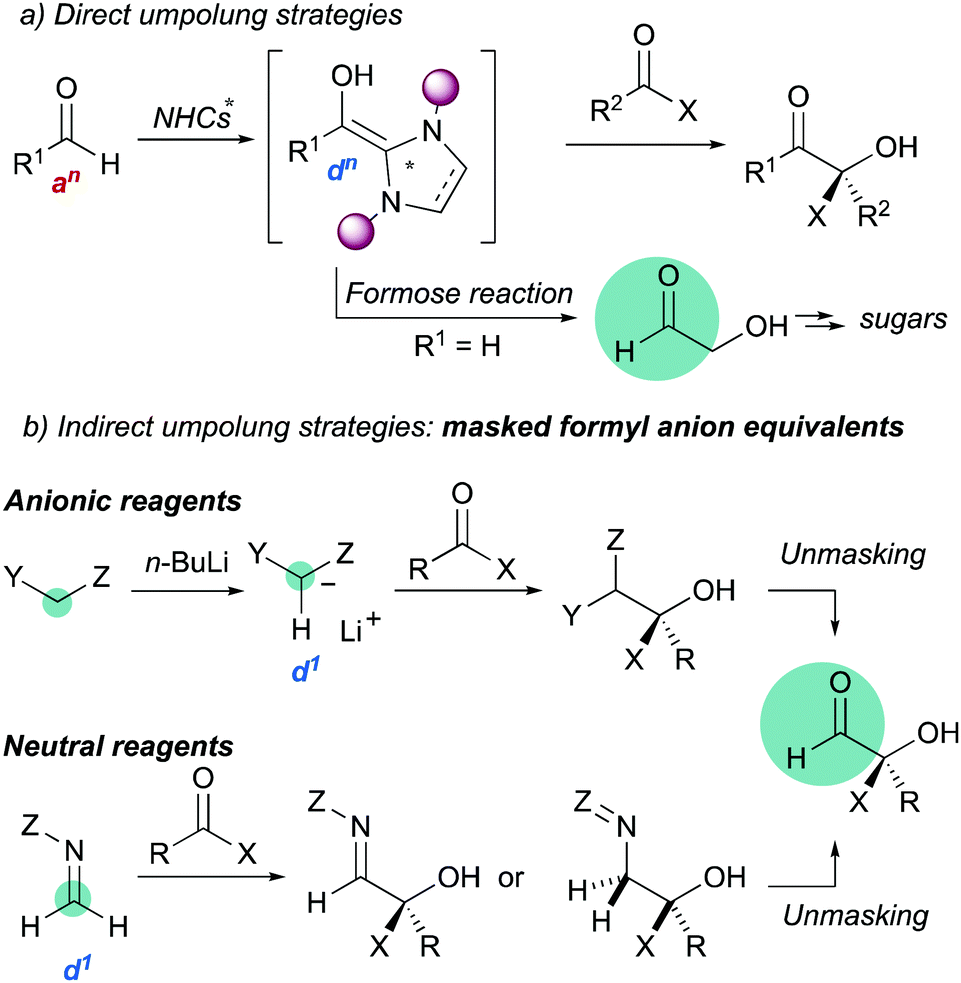
Umpolung strategies for inverting the natural reactivity of functional groups are powerful synthetic tools for generation of molecular complexity in organic synthesis.1 In particular, the asymmetric 1,2-addition of acyl anion equivalents (d1 reagents) to carbonyl compounds provides direct access to valuable functionalized alcohols.2 Most direct nucleophilic acylations are catalyzed by N-heterocyclic carbenes (Scheme 1a),3 but this strategy fails in the case of formaldehyde due to oligomerizations (the formose reaction).4 Therefore, several masked formyl anion reagents (anionic or neutral) have been developed to circumvent such limitation (Scheme 1b).5 These strategies are based on two key steps: (i) asymmetric C–C bond formation, and (ii) subsequent unmasking to reveal the formyl group.
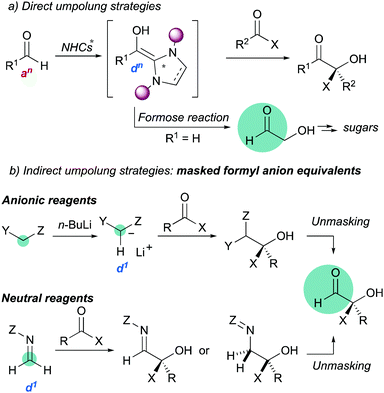 |
| | Scheme 1 Carbonyl umpolung strategies. | |
Approaches based on anionic reagents require in general harsh, strongly basic conditions and catalytic asymmetric variants have not been developed. Whereas this feature article will focus on the use of the more versatile neutral formyl anion equivalents in asymmetric catalysis, a selection of chiral auxiliary-based reagents is also shown, with the aim to provide a clear background of the topic. It should be noted that, for some reagents, the catalytic systems are highly reminiscent of the corresponding stoichiometric methods that inspired them and for others, the catalytic enantioselective reactions remain elusive. Key aspects which have allowed us to develop efficient reactions employing formaldehyde tert-butyl hydrazone (FTBH) as masked d1 reagent in combination with bifunctional organocatalysts are highlighted.
2. Background: from chiral auxiliary-based reagents to enantioselective reactions
2.1. Anion-stabilized reagents
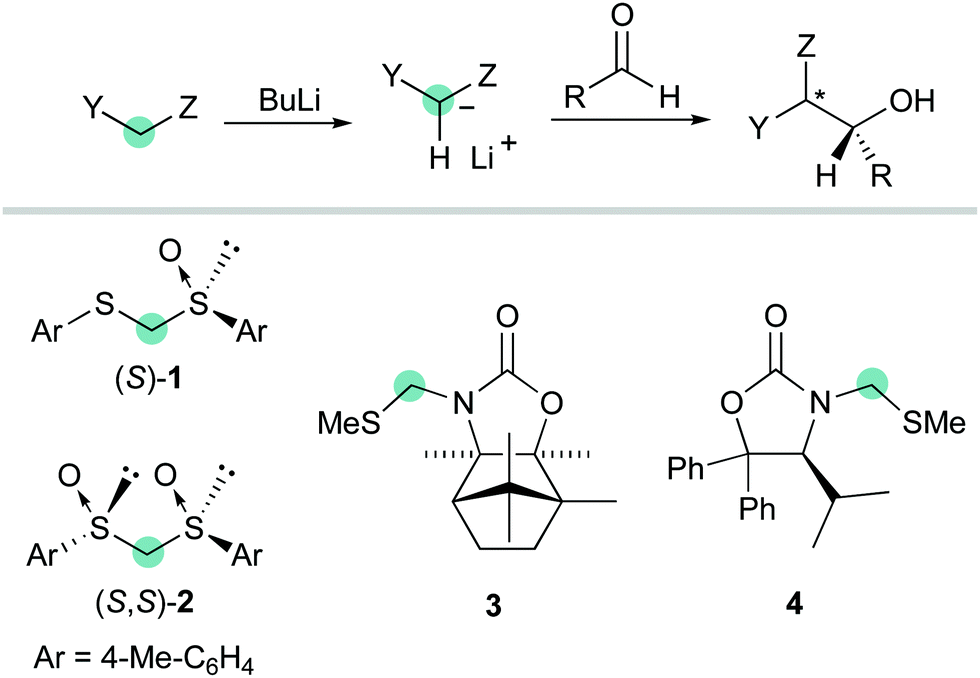
A number of studies have been focused on diastereoselective additions to aldehydes accomplished by using sulfur-stabilized carbanions carrying various chiral auxiliaries such as dithioacetal mono-S-oxide (S)-1,6C2-symmetric bis-sulfoxide (S,S)-2,7 as well as camphor- or valine-derived oxazolidinone S,N-acetals 38 and 49 (Scheme 2).
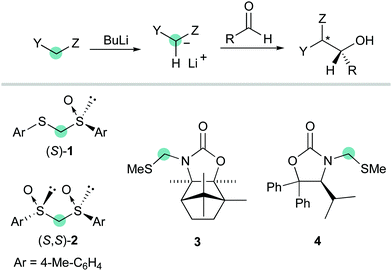 |
| | Scheme 2 Chiral auxiliary-based anionic formyl anion equivalents. | |
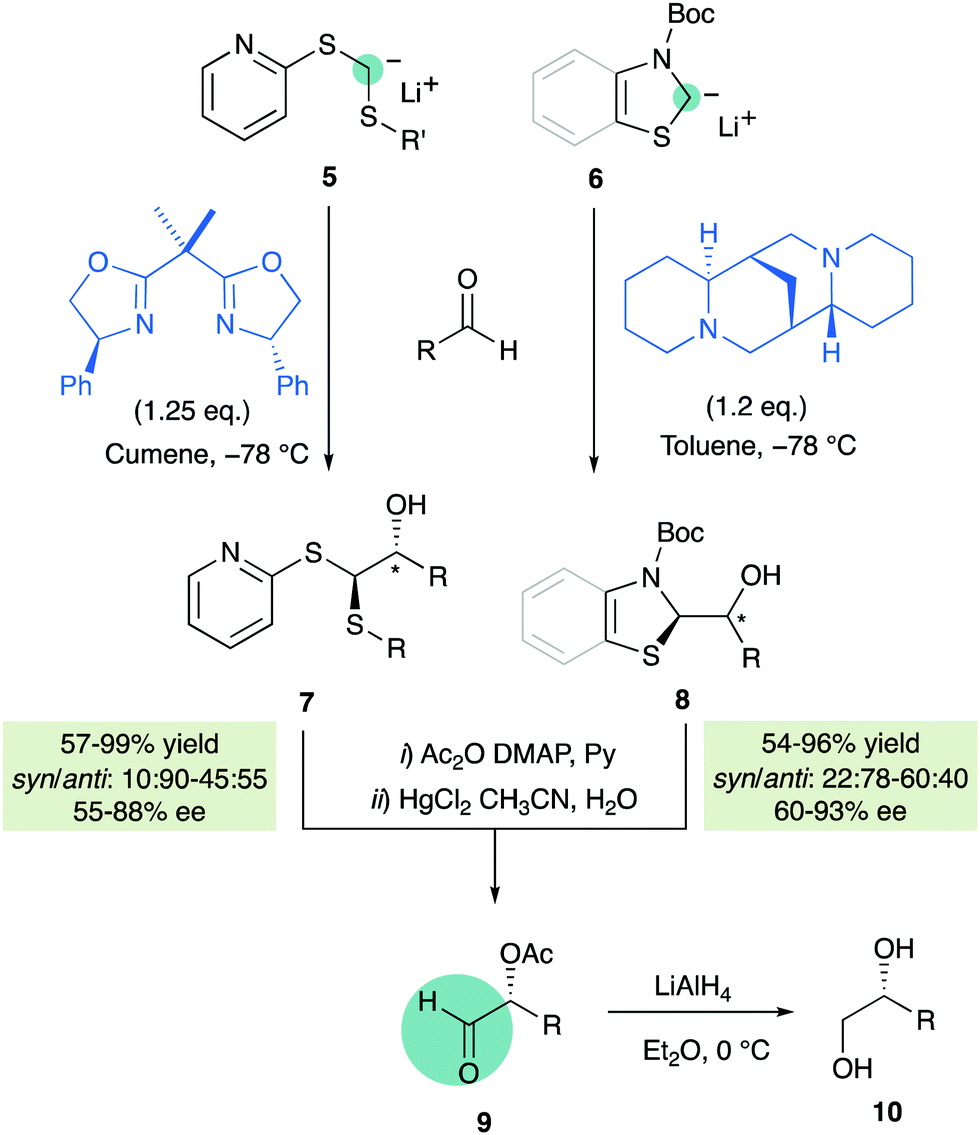
Exploiting again the carbanion stabilizing properties of sulfur, Toru and co-workers reported in 2004 some non-catalytic enantioselective approaches employing achiral α-lithiated dithioacetals 510 or N-Boc-thiazoline/benzothiazoline 611 in the presence of stoichiometric amounts of enantiopure additives such as bis(oxazolines) or (−)sparteine, respectively (Scheme 3). Acetylation of the primary adducts followed by treatment with mercury(II) chloride afforded aldehydes 9 which were directly transformed into enantiomerically enriched diols 10.
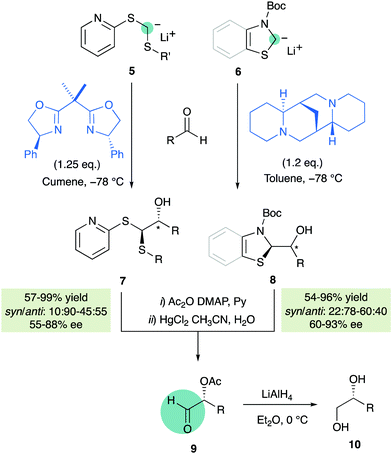 |
| | Scheme 3 Non-catalytic enantioselective approaches for nucleophilic formylations. | |
As mentioned before, however, the development of catalytic enantioselective versions of the above-mentioned methodologies have been hampered by the strongly basic character of such anionic reagents, incompatible with many catalysts and functional groups, that underwent undesired side reactions. Moreover, unmasking of the formyl group from the primary adducts is still a major difficulty, requiring toxic mercury salts, iodine reagents or multistep sequences.
2.2. Neutral reagents: N,N-dialkyl hydrazones
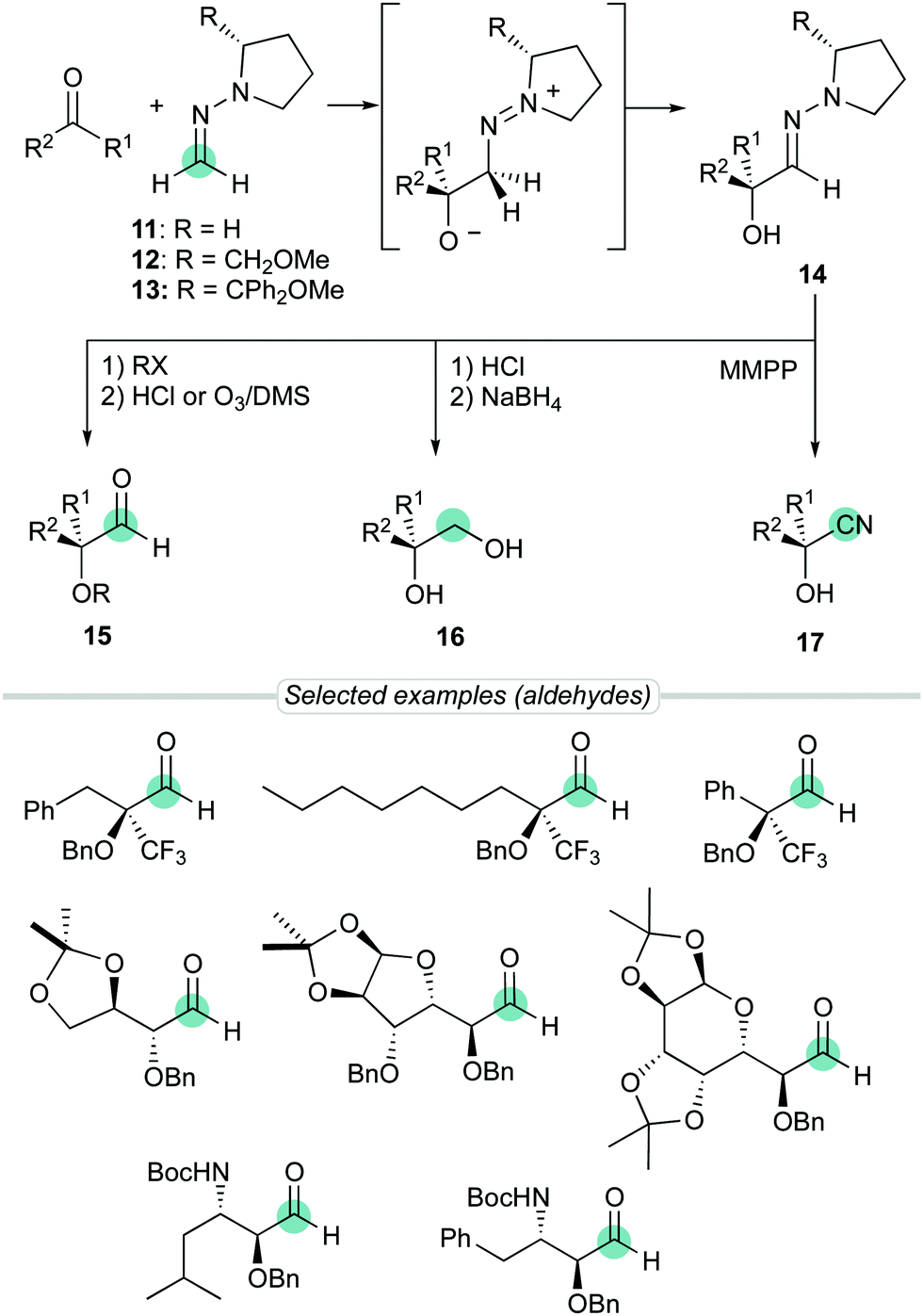
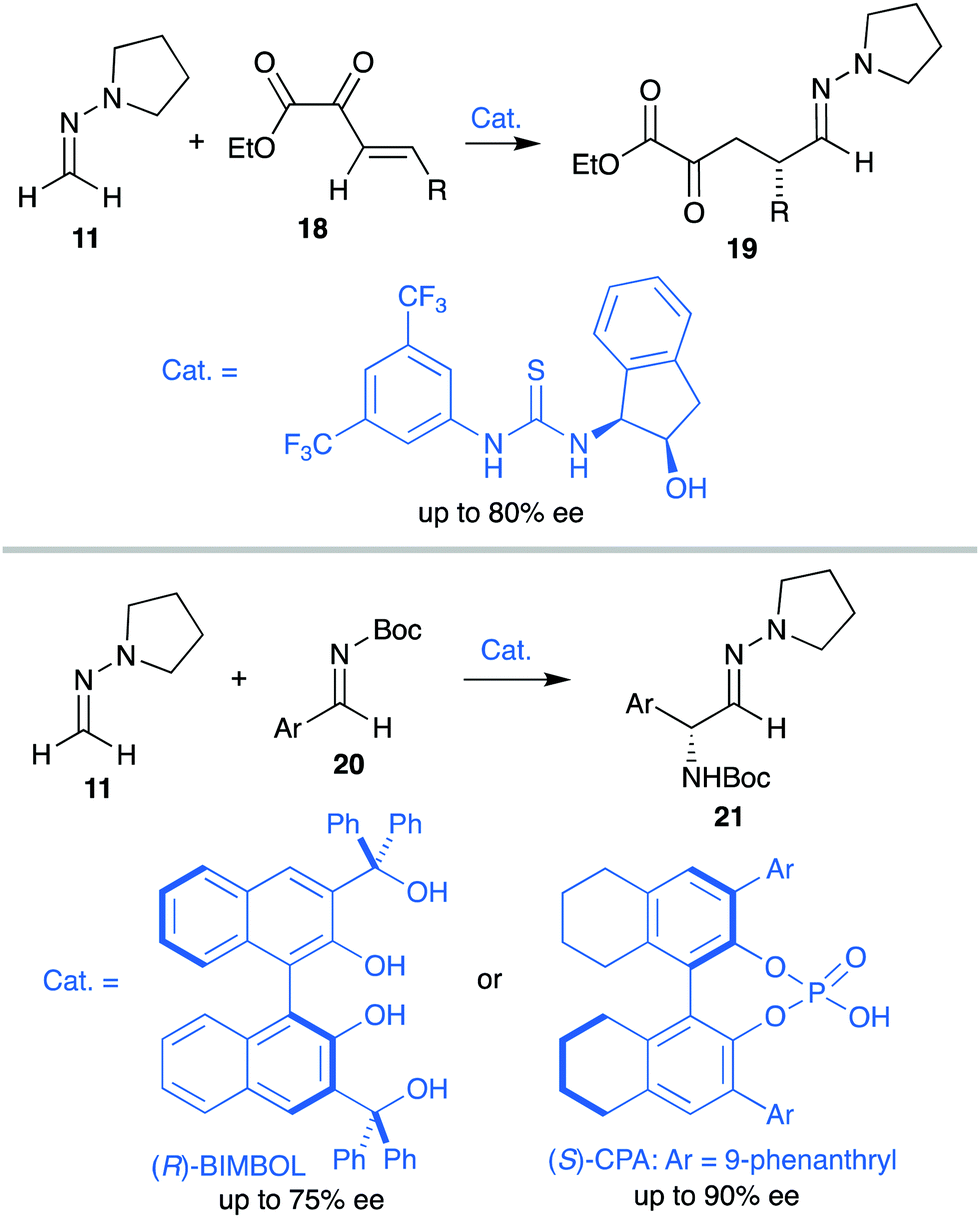
Over the years, we have exploited the enhanced aza-enamine (nucleophilic) character of formaldehyde N,N-dialkyl hydrazones for the stereoselective introduction of single-carbon functional groups into more complex molecular scaffolds.12 In particular, the enhanced reactivity provided by the pyrrolidine ring was exploited to perform diastereoselective additions to diverse electrophiles, including carbonyl compounds. Thus, the simplest achiral derivative 11, along with chiral proline derivatives such as SAMP [(S)-1-amino-2(methoxymethyl)pyrrolidine]hydrazone 1213 or analogue 13 have been successfully employed in reactions with chiral α-alkoxy- and α-amino aldehydes14 (substrate-controlled stereoselectivity) and simple aldehydes15 or trifluoromethyl ketones16 (reagent-controlled stereoselectivity) to afford densely functionalized α-hydroxy hydrazones 14 (Scheme 4). These procedures, based in a soft and neutral reagent, not only benefit of milder reaction conditions, but also provides a remarkable versatility associated to efficient transformations of the hydrazone moiety. The primary targets, aldehydes 15, can be readily synthesized by C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N bond cleavage using acid hydrolysis or ozonolysis,17 and eventually reduced to diols 16. Alternatively, cyanohydrin derivatives 17 can also be easily obtained after oxidative N–N bond cleavage (aza-Cope type elimination) promoted by magnesium monoperoxyphthalate hexahydrate (MMPP).18
N bond cleavage using acid hydrolysis or ozonolysis,17 and eventually reduced to diols 16. Alternatively, cyanohydrin derivatives 17 can also be easily obtained after oxidative N–N bond cleavage (aza-Cope type elimination) promoted by magnesium monoperoxyphthalate hexahydrate (MMPP).18
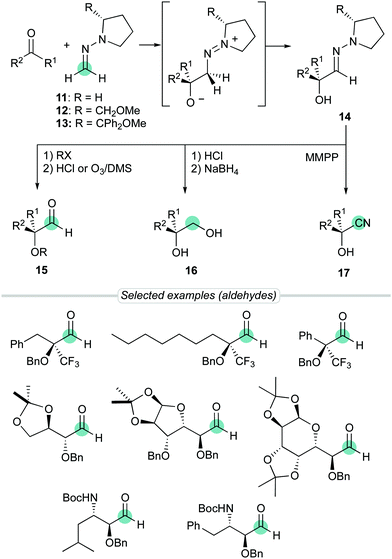 |
| | Scheme 4 Chiral auxiliary-based formaldehyde N,N-dialkyl hydrazones as neutral masked formyl anion equivalents. | |
The development of catalytic enantioselective versions of these reactions proved to be a difficult task due to the sensitivity of hydrazones toward most Lewis acidic metal complexes, usually employed for the activation of weakly electrophilic substrates.19 The milder nature of organocatalytic activation strategies, however, appeared to solve these compatibility issues.20 Thus, H-bonding activation by thioureas was identified as a suitable strategy for the conjugate addition of achiral pyrrolidine derivative 11 to β,γ-unsaturated α-ketoesters 18,21 while LUMO-lowering activation by axially chiral BINOL, phosphoric (CPA) or dicarboxylic acid derivatives allowed 1,2-addition of 11 to N-Boc protected imines 20 (Scheme 5).22 The corresponding products 19 and 21, respectively, were obtained with moderate average enantioselectivities in both cases, underlining the need of more efficient activation modes. Moreover, none of these strategies provided satisfactory results in 1,2-additions of N,N-dialkyl hydrazones to carbonyl compounds. These limitations prompted us to explore a dual activation mode as an alternative strategy for the development of the targeted enantioselective formylation of these types of substrates.
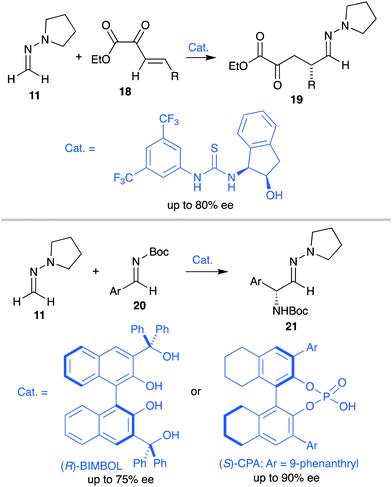 |
| | Scheme 5 Seminal enantioselective organocatalytic reactions employing formaldehyde N,N-dialkyl hydrazones. | |
3. Asymmetric organocatalytic 1,2-additions of formaldehyde tert-butyl hydrazone to carbonyl compounds
3.1. Hypothesis and design of the catalytic system
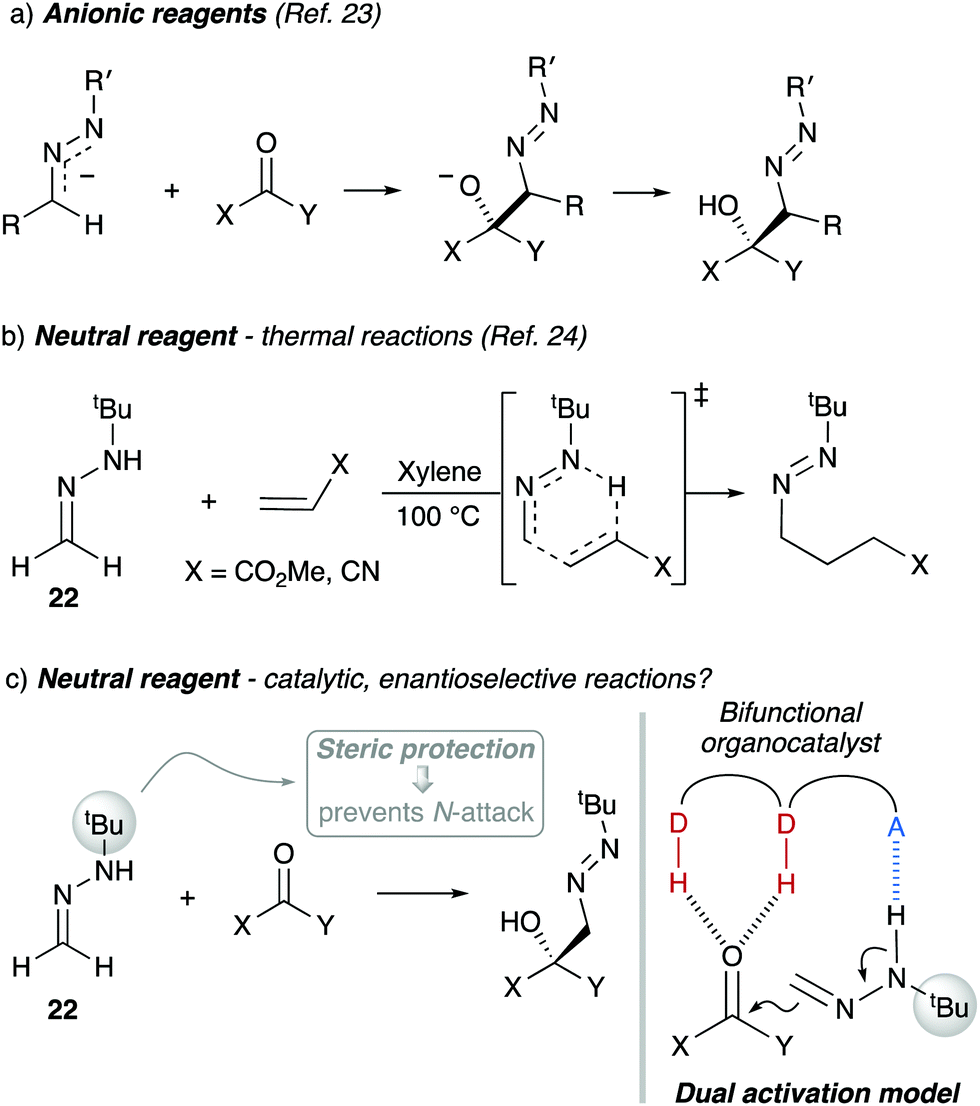
Due to the N,N-disubstitution in hydrazones such as 11, the attack of the nitrogen atom to neutral electrophilic reagents results in the formation of zwitterionic compounds in a non-productive, reversible process. In monosubstituted analogues, however, the attack of the more nucleophilic nitrogen atom must be avoided to prevent undesired side reactions. In this context, Baldwin and co-workers had reported on the use of azo-anions from N-tert-butyl hydrazones as acyl anion equivalents using the steric protection provided by the bulky tert-butyl group to avoid reaction at the N atom (Scheme 6a), although the reaction with the formaldehyde derivative was not reported.23 Later, the same group also reported on the reactivity of neutral N-monosubstituted hydrazones in thermal ene reactions with highly reactive Michael acceptors such as methyl acrylate and acrylonitrile (Scheme 6b).24 Inspired by these pioneering reports, we envisioned that reactions of neutral formaldehyde N-tert-butyl hydrazone (FTBH) 22 with carbonyl compounds should take place at the azomethine carbon for steric reasons, while the presence of the alkylamino NH group could eventually offer additional opportunities for interactions with bifunctional organocatalysts (Scheme 6c). This strategy proved to be successful for 1,2-additions to carbonyl compounds (formally hetero-carbonyl-ene reactions) as disclosed below.
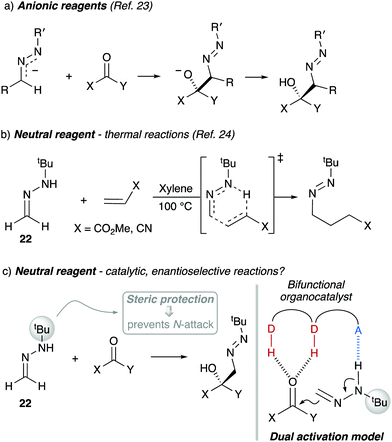 |
| | Scheme 6 Dual activation strategy with formaldehyde N-tert-butyl hydrazone 22. D–H: H-bond donor, A: H-bond acceptor. | |
3.2. Addition of FTBH to activated carbonyl compounds
The first asymmetric organocatalytic reactions were planned considering a series of carbonyl substrates containing additional activating functionalities such as a second carbonyl group in α-keto esters25 and isatins (1,2-dicarbonyl systems)26 or a phosphoryl group in α-keto phosphonates.27 In this way, well-defined three-dimensional environments are expected to be created upon engagement with H-bond donor catalysts by multiple-point interactions.
α-Keto esters.
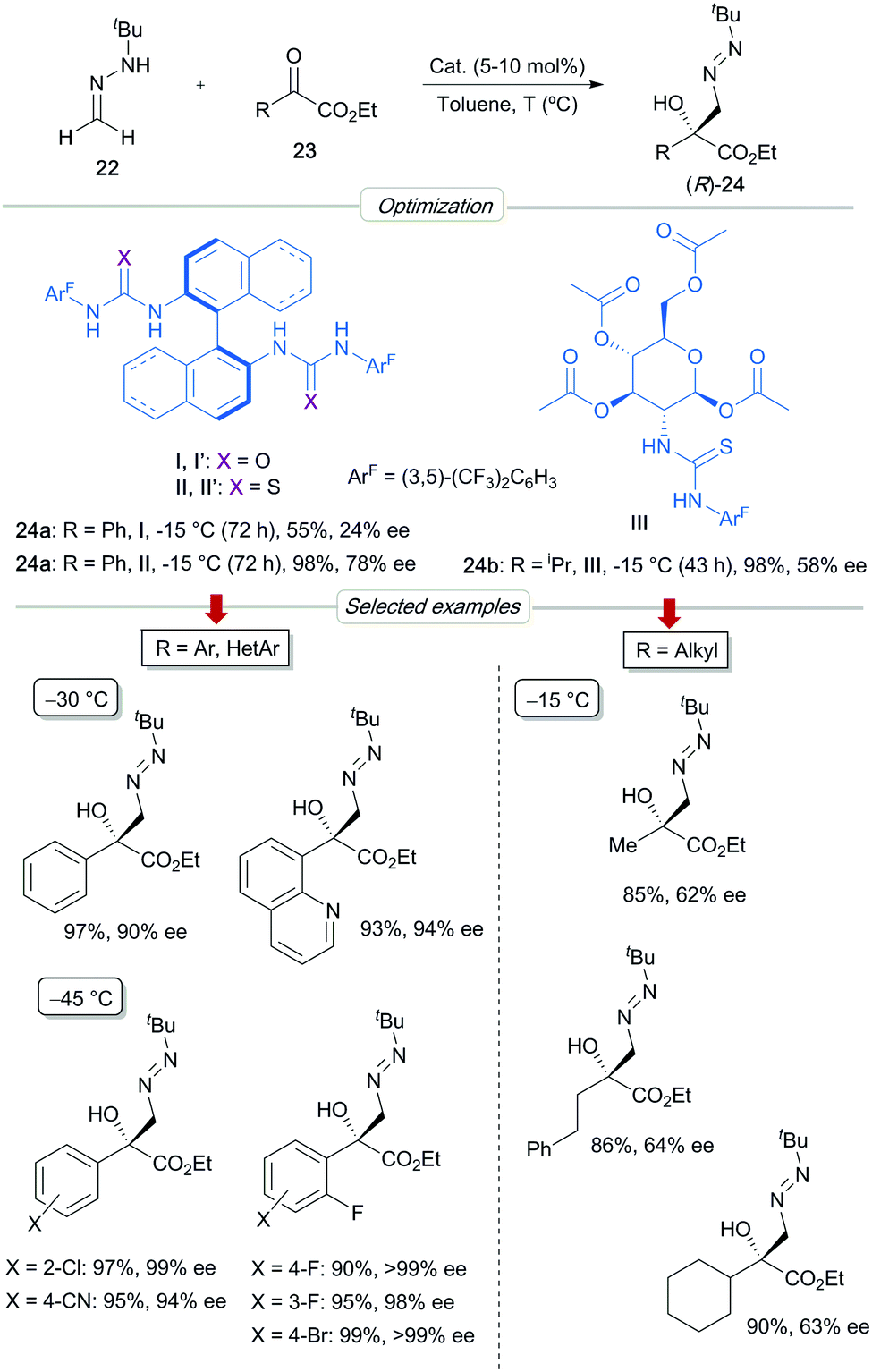
Ethyl phenylglyoxylate 23a was initially chosen as a model α-keto ester substrate (Scheme 7). The preliminary screenings served to identify BINAM-bis urea I as the best catalyst, which afforded, in toluene at −15 °C, the expected azomethyl alcohol 24a in 98% yield and 78% ee. Remarkably, bis-thiourea analogue II proved to be a less efficient catalyst (55% yield), leading to the product 24a with a low 24% ee and with the opposite sense of enantioinduction. This a priori anomalous divergence suggests different activation modes for both catalysts. The enantioselectivity was further improved to 90% ee by performing the reaction at −30 °C, without compromising the chemical yield. The scope of the reaction was demonstrated with a range of aromatic and heteroaromatic substrates. Aryl-substituted azomethine alcohols 24 were obtained in high yields and enantioselectivities (up to >99% ee), either employing BINAM (I) or H8-BINAM (I′) bis-urea catalysts. The superior reactivity of electron-poor derivatives made it possible to perform reactions at −45 °C, reaching excellent enantioselectivities in most cases (94–99% ee). Remarkably, essentially pure enantiomers (R)-24 were regularly obtained for ortho-fluorinated derivatives. Alkyl-substituted α-keto esters proved to be more challenging substrates for which enantioselectivities were highly dependent on the alkyl chains. For most of these substrates, multifunctional D-glucosamine-derived thiourea III showed slightly superior catalytic activity than (R)-BINAM bis-urea I, reaching full conversions in cleaner reactions, albeit with moderate enantioselectivities (up to 64% ee).
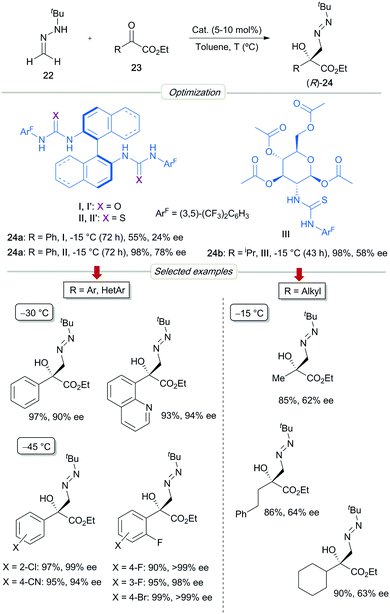 |
| | Scheme 7 Asymmetric addition of FTBH to α-keto esters. | |
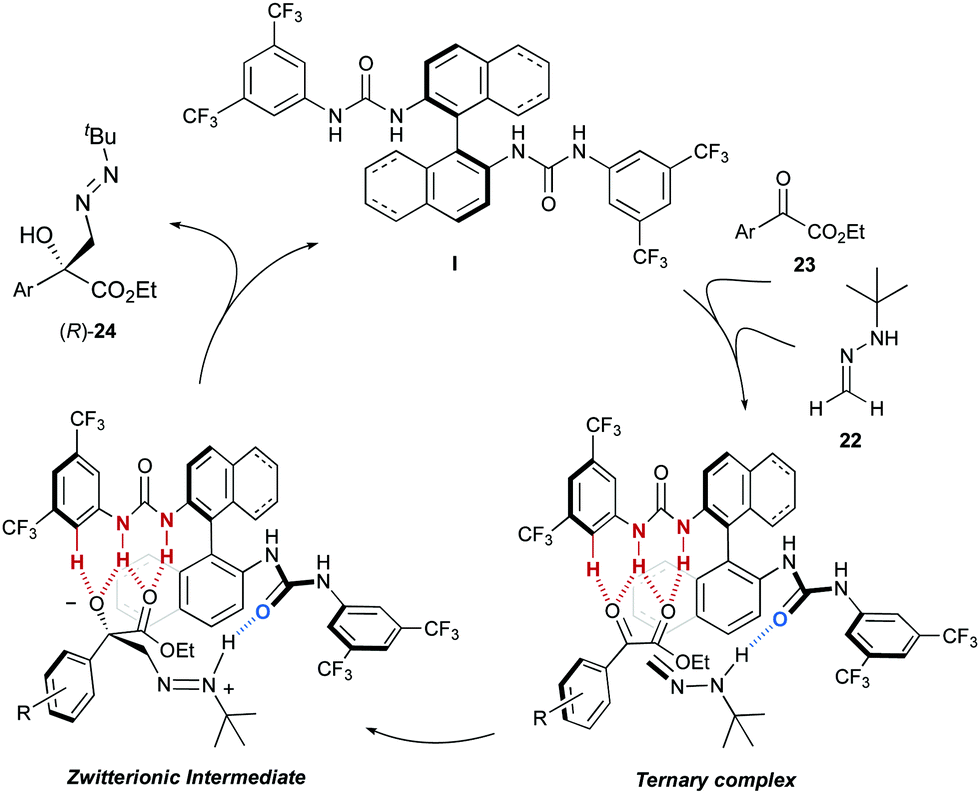
Considering the close structural similarity between bis-urea I (I′) and bis-thiourea II (II′) catalysts, the higher hydrogen bond acceptor capability of the oxygen in the carbonyl group of the urea was suggested as the distinct factor. Accordingly, the mechanism and stereochemical model depicted in Scheme 8 was proposed. Hence, a bifunctional behaviour of the catalyst results in the activation of both the hydrazone, by means of a NH⋯OC hydrogen bond, and the α-keto ester, through a multiple hydrogen bond network provided by a second urea unit. In this way, a ternary complex is generated, in which the orientation of the bidentate electrophile avoids the aromatic ring being placed in the inner, more crowded region of the catalyst. After nucleophilic attack of the azomethine carbon to the activated carbonyl group and release of the product, the model predicts the formation of the product with the observed (R) absolute configuration at the newly created stereogenic center.
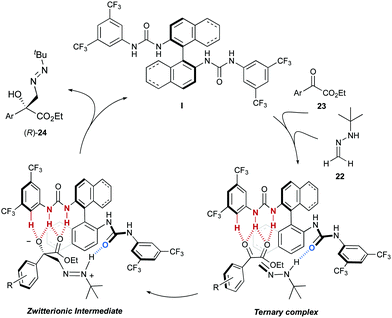 |
| | Scheme 8 Proposed catalytic cycle and stereochemical model. | |
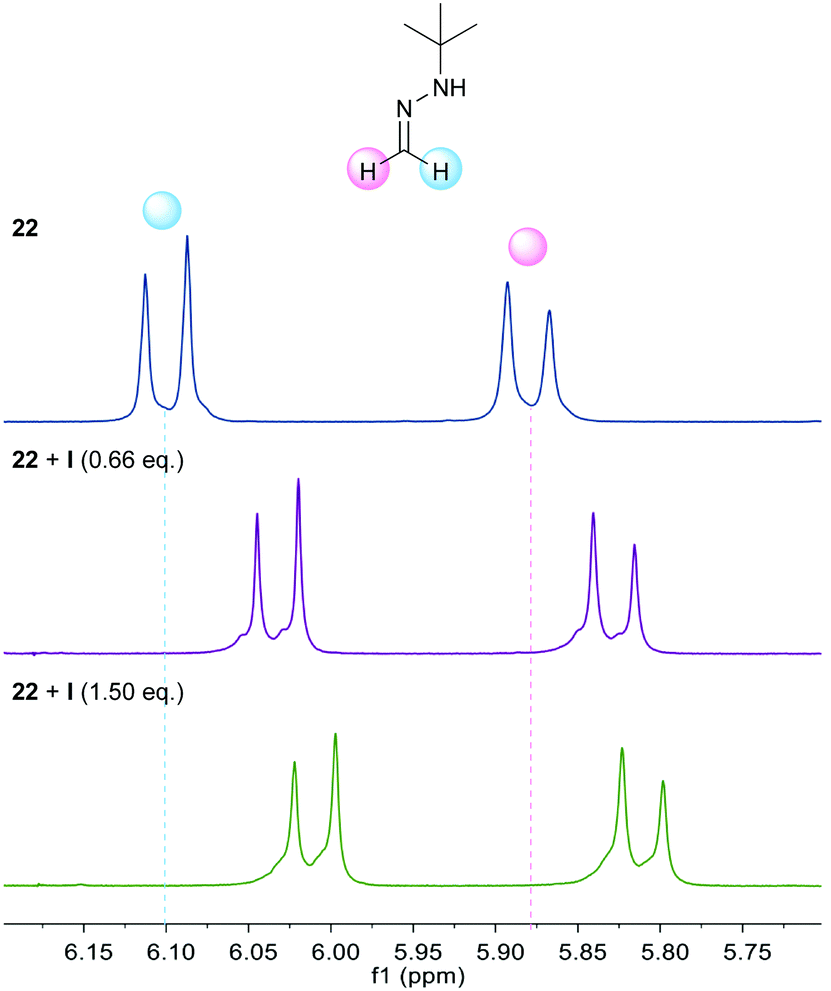
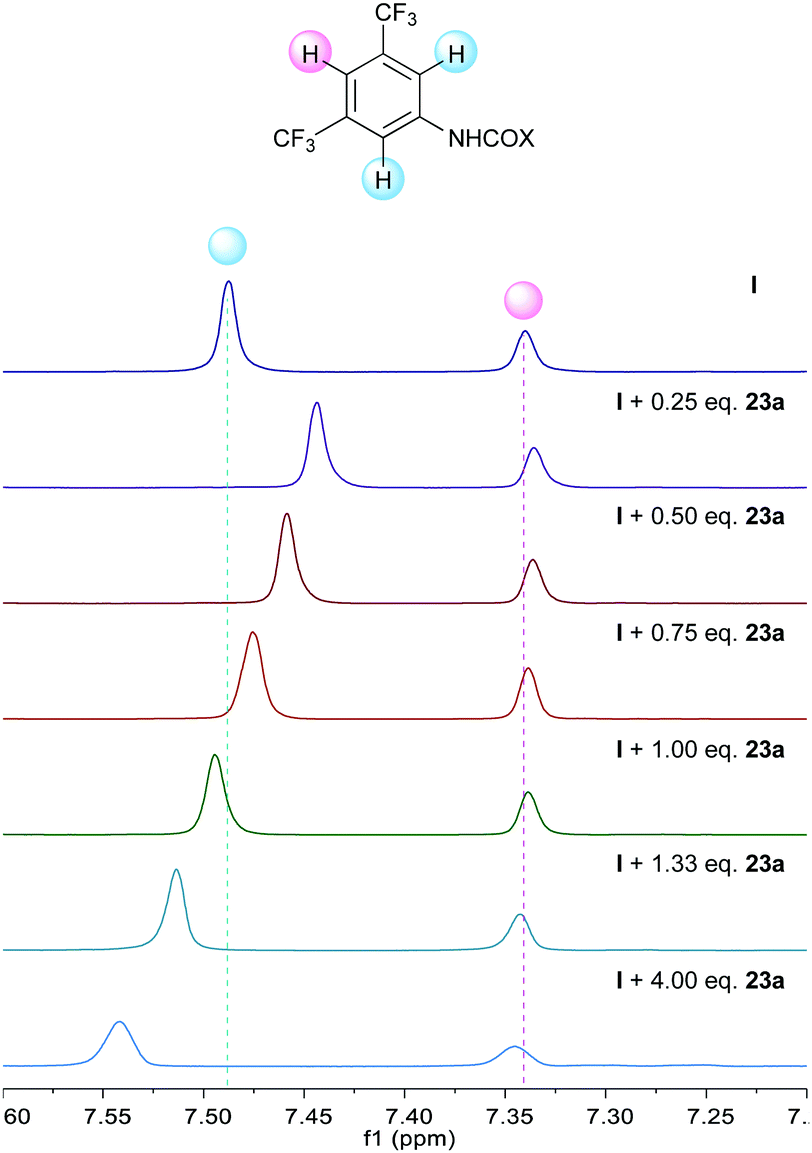
Additional support was obtained from NMR experiments. Thus, 1H NMR spectra recorded for 22 in the presence of increasing amounts of catalyst I showed the signals of the azomethine protons shifted upfield (Δδ up to −0.1 ppm at a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio, Fig. 1), as expected from a higher electron density at the azomethine carbon resulting from the polarization of the N–H bond. Moreover, 1H NMR spectra recorded for I in the presence of increasing amounts of keto ester 23a showed an additional perturbation of the ortho-protons of the catalyst (Fig. 2), suggesting their participation as H-bond donors in a cooperative network.28 Initially, an upfield shift of the signals of these protons was observed upon addition of 0.25 equiv. of keto ester, presumably due to the disruption of inter- and/or intra-molecular self-aggregation of bis-urea catalyst. Progressively, a downfield shift was then observed upon further addition of 23a (Δδ up to 0.1 ppm at a 1:4 ratio), consistent with the establishment of a catalyst–substrate intermolecular H-bonding network.
:1 ratio, Fig. 1), as expected from a higher electron density at the azomethine carbon resulting from the polarization of the N–H bond. Moreover, 1H NMR spectra recorded for I in the presence of increasing amounts of keto ester 23a showed an additional perturbation of the ortho-protons of the catalyst (Fig. 2), suggesting their participation as H-bond donors in a cooperative network.28 Initially, an upfield shift of the signals of these protons was observed upon addition of 0.25 equiv. of keto ester, presumably due to the disruption of inter- and/or intra-molecular self-aggregation of bis-urea catalyst. Progressively, a downfield shift was then observed upon further addition of 23a (Δδ up to 0.1 ppm at a 1:4 ratio), consistent with the establishment of a catalyst–substrate intermolecular H-bonding network.
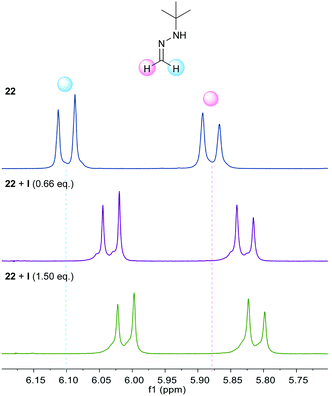 |
| | Fig. 1
1H NMR titration experiment of 22 with catalyst I. | |
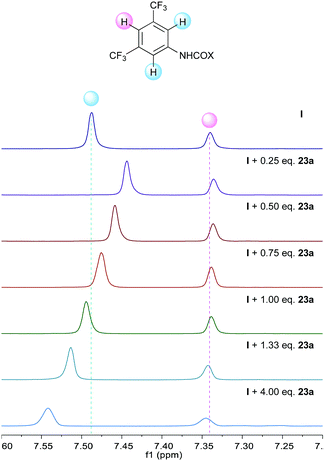 |
| | Fig. 2
1H NMR titration experiment of I with α-keto ester 23a. | |
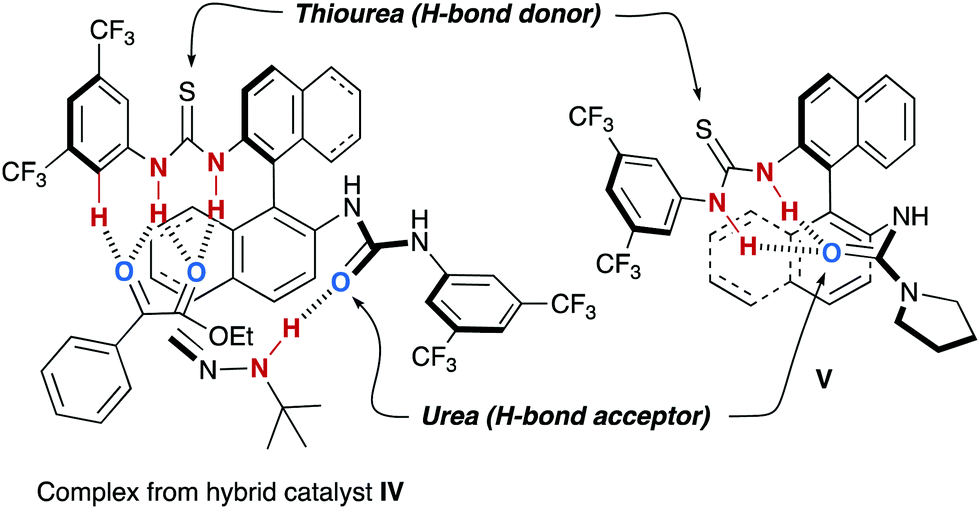
Further support for the proposed bifunctional working model was collected from experiments performed with hybrid thiourea–urea catalyst IV (Fig. 3). According to the proposed mechanism, a single urea moiety is required to activate the reagent 22 and, therefore, IV was expected to imitate the behaviour of I, and not that of II. In fact, this hybrid catalyst afforded azomethyl alcohols with similar enantiomeric excess as I and slightly shorter reaction times, which accounts for the better activation of α-keto esters by the thiourea moiety. However, there is a limitation on the catalyst design: incorporation of a better H-bond acceptor urea, as in pyrrolidine derivative V, results in a lower catalytic activity and the obtention of racemic products. These facts can be explained by the deactivation of the catalyst due to a strong intramolecular H-bonding interaction between the amide carbonyl group, with enhanced H-bond acceptor ability, and the thiourea moiety as a double H-bond donor.29
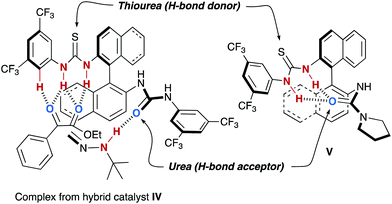 |
| | Fig. 3 Inter or intramolecular H-bonding networks with hybrid thiourea–urea catalysts IV and V. | |
Diazenes 24 are densely functionalized tertiary alcohols which contain the core structures of pharmacologically relevant compounds such as Anisodine, Voriconazole (VFEND®),30 Posaconazole (NOXAFIL®),31 and isoserines (2-substituted α-hydroxy-β-amino acids) which are present in several taxoid-based anticancer agents,32 among others.
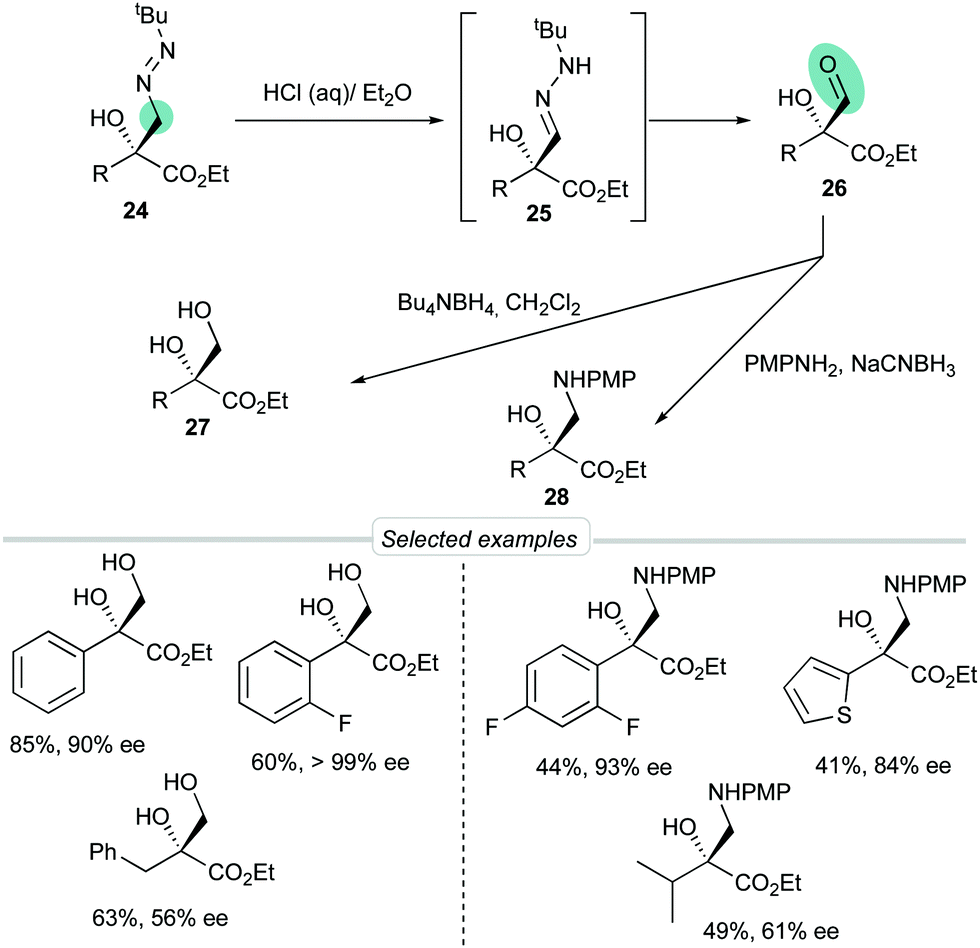
To demonstrate that FTBH 22 can indeed be designated as a formyl anion equivalent, representative diazenes 24 were transformed into aldehydes 26 through a tautomerization (→ 25)/hydrolysis sequence efficiently performed by simple treatment with HCl in a biphasic H2O/Et2O medium (Scheme 9). Crude aldehydes were sensitive products that could not be purified by chromatographic techniques but were isolated with a high degree of purity and could be used directly in subsequent transformations. For example, reductions or reductive aminations afforded diols 27 or α-hydroxy-β-amino esters 28 in good overall yields.
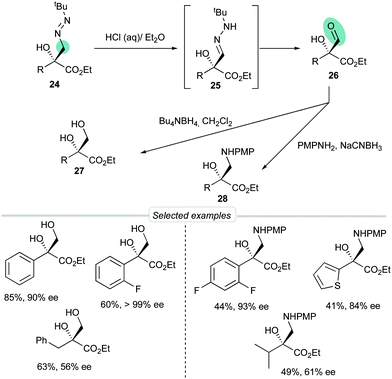 |
| | Scheme 9 Unmasking by hydrolysis and further transformations. | |
Isatins.
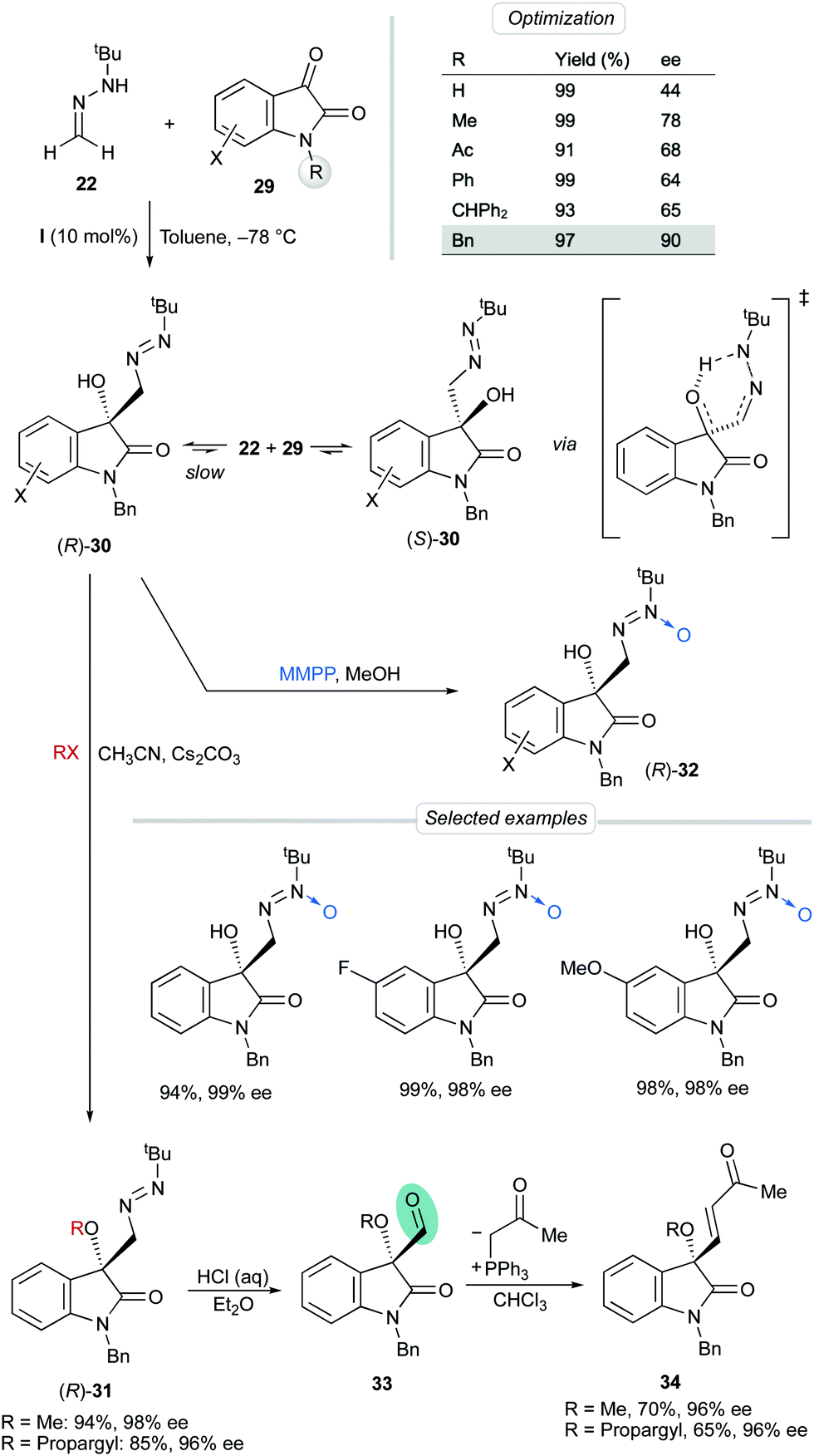
Aiming to expand the scope of this dual activation strategy, we reported later the enantioselective reaction of 22 with isatins 29, a different family of dicarbonyl substrates. The particular motivation in this case was the construction of the important 3-hydroxy 2-oxindol core, found in a lot of natural products and plenty of bioactive molecules.33 The influence of the substitution at the amide nitrogen atom was investigated using bis-urea catalyst I in toluene at −78 °C. N-Benzyl derivative 29 (X = H, R = Bn) afforded the desired adduct 30 in high yield and the highest enantioselectivity (90% ee). Unfortunately, though, such high enantioselectivities were observed only in isolated experiments, and, on the other hand, it was found out that enantiomerically enriched samples slowly racemized in solution, even at low temperatures. The uncatalyzed racemization is believed to proceed by a thermal retro-hetero-carbonyl ene reaction, through a 6-membered ring transition state involving an intramolecular OH⋯N bond (Scheme 10). Fortunately, however, in situ O-alkylation of 30 regularly afforded O-methyl and O-propargyl derivatives 31 in excellent yields and enantioselectivities (96–98% ee). Alternatively, rapid oxidation of the fresh crude diazenes was performed with magnesium monoperoxyphthalate hexahydrate (MMPP·6H2O) to afford azoxy compounds 32 in high yields, complete regioselectivities and excellent enantioselectivities (up to 99% ee).34 As anticipated, O-alkylated products and azoxy compounds were configurationally stable, as the hypothesized racemization mechanism is avoided in both cases. The diazene-to-aldehyde transformation from products 31 was easily performed under acidic hydrolysis and the crude aldehydes 33 were subsequently used in Wittig olefinations to give enones 34 in good overall yields.
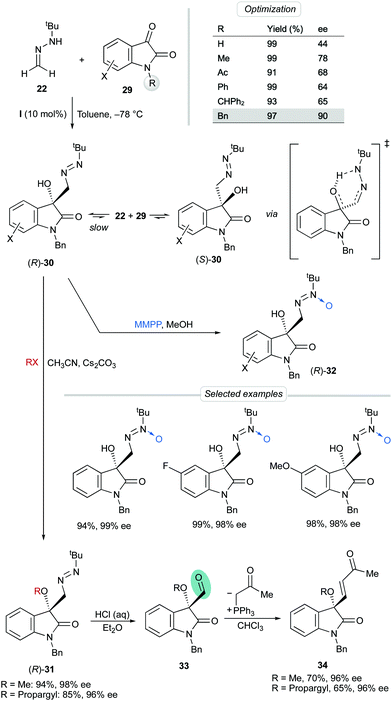 |
| | Scheme 10 Asymmetric addition of FTBH to isatins. | |
α-Keto phosphonates.
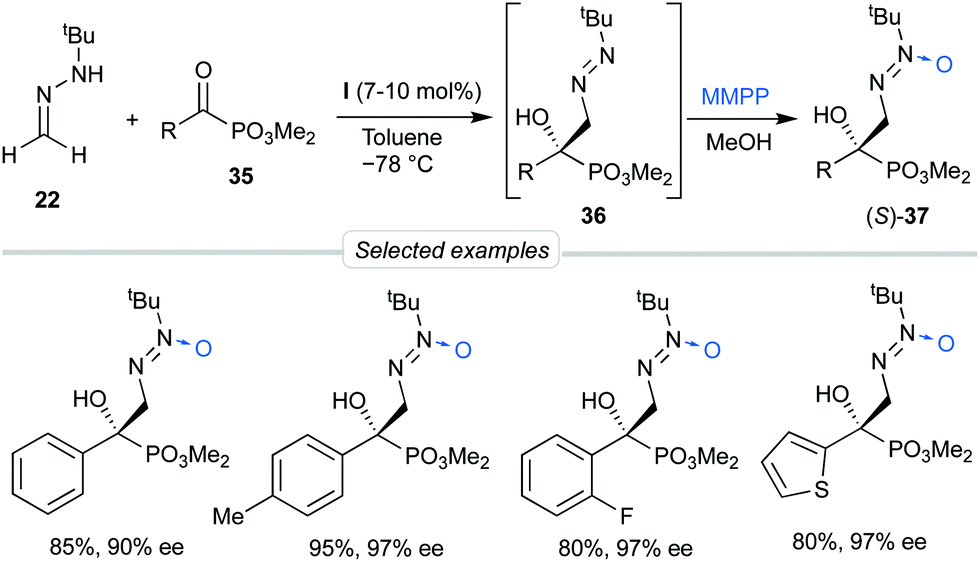
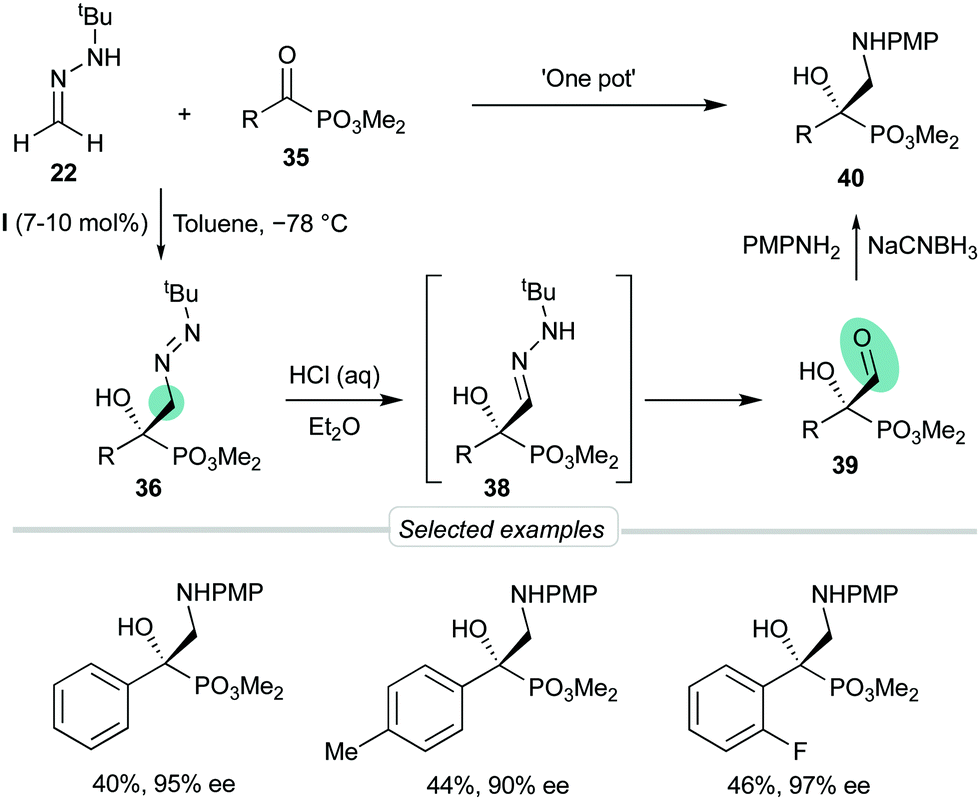
Considering the possibility of incorporating other substrates into the dual activation model by bis-urea catalyst I, we next decided to explore the behavior of α-keto phosphonates 35 as activated electrophiles, aiming to obtain functionalized α-aryl α-hydroxy phosphonates, precursors of biologically active phosphaisoserines.35 The phosphonate is a strong electron-withdrawing group which, unlike planar carbonyl derivatives, is featured by a tetrahedral phosphorous centre next to the reactive carbonyl group. These substrates showed high reactivities in absence of catalysts, making necessary to control the undesired background reaction. Moreover, diazenes 36 were relatively unstable and a ‘one-pot’ oxidation into azoxy compounds 37 was required to analyze the reaction by chromatographic techniques (Scheme 11). To our delight, employing again BINAM bis-urea I in toluene at −78 °C, this procedure afforded azoxy compounds 37 in good overall yields and excellent enantioselectivities (up to 97% ee). Transformation of the N-tert-butyldiazenylmethyl group of crude products 36 into the formyl group in aldehydes 39 was also performed by treatment with HCl in a biphasic H2O/Et2O medium via hydrazone intermediates 38. Aldehydes 39 were not isolable by chromatographic techniques but were directly subjected to reductive amination to afford quaternary β-amino-α-hydroxyphosphonates 40 in satisfactory overall yields (3 reactions, 1 chromatographic purification) and high enantioselectivities (Scheme 12).
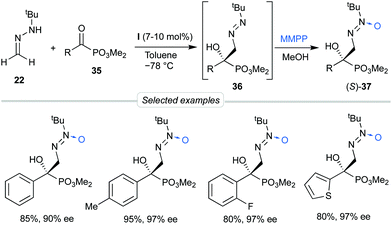 |
| | Scheme 11 Asymmetric addition of FTBH to α-keto phosphonates. | |
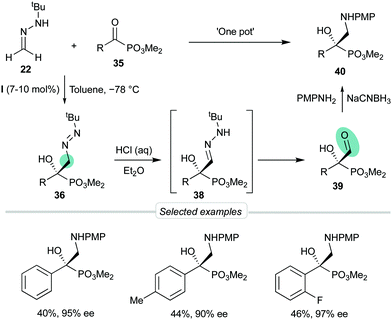 |
| | Scheme 12 Synthetic sequence to β-amino-α-hydroxyphosphonates. | |
3.3. Addition of FTBH to monocarbonyl compounds
In the next stage, we investigated enantioselective reactions of FTBH with single carbonyl compounds (simple aldehydes36 and fluoromethyl ketones37). These are a priori more challenging substrates for two main reasons: first, the intrinsic electrophilicity of the carbonyl group in these substrates, highly dependent on the substitution patterns, is expected to translate into a significant uncatalyzed, racemic background reaction. Second, the geometry of the catalyst–substrate complexes should be fixed using weak H-bonding interactions with a single acceptor site, in contrast with the above-mentioned precedents.
Simple aldehydes.
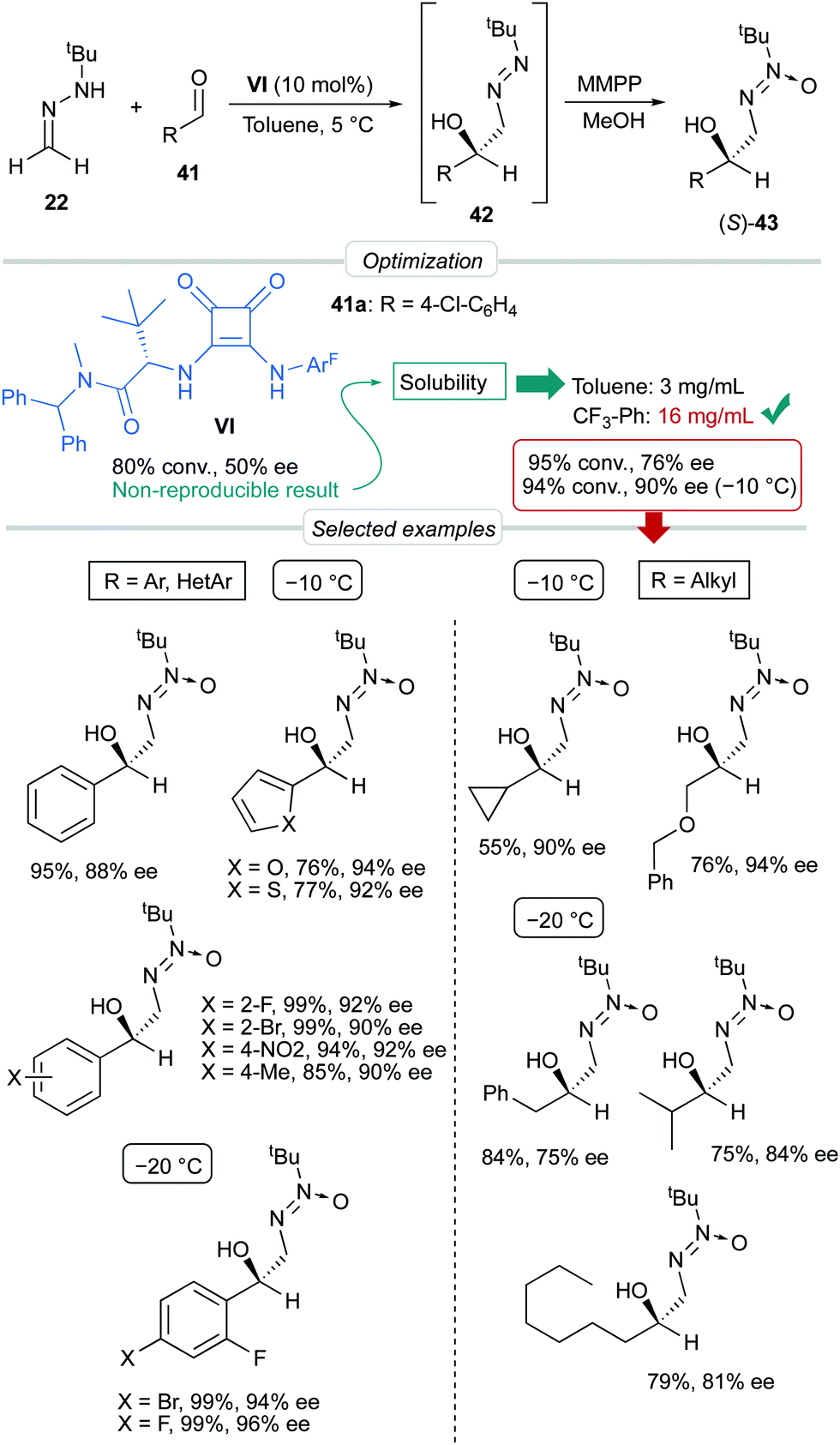
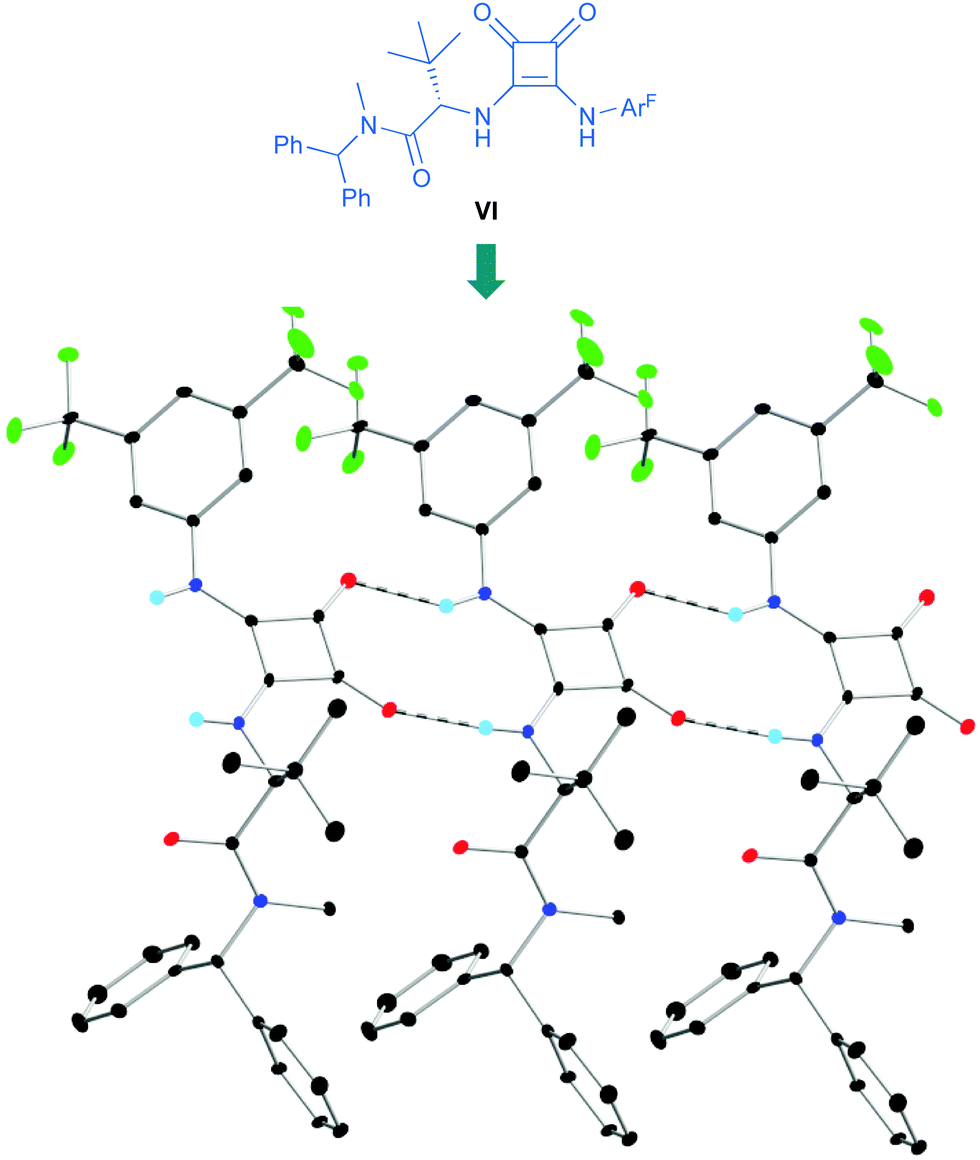
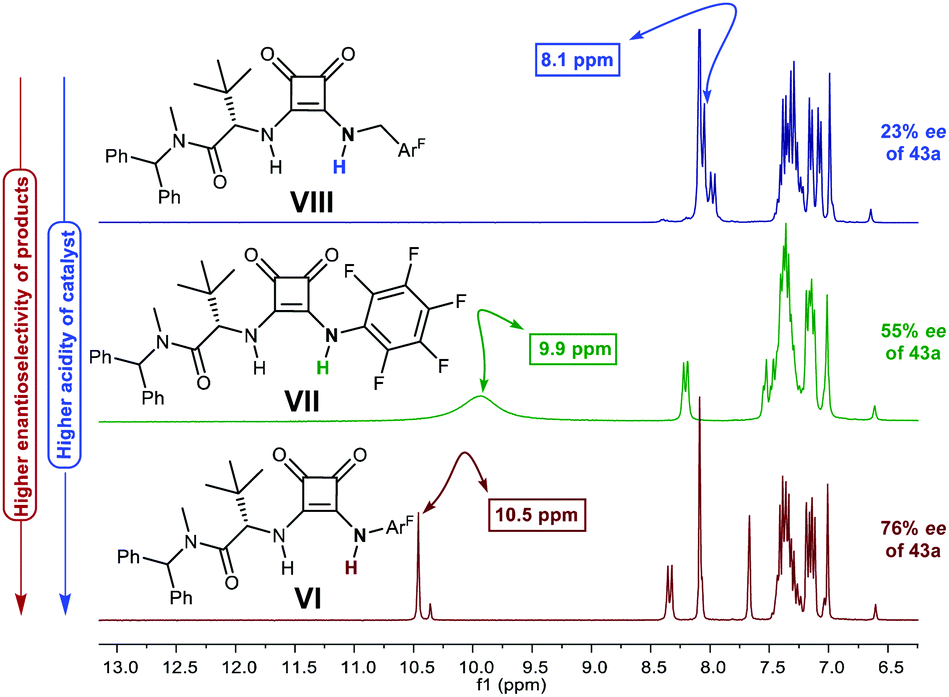
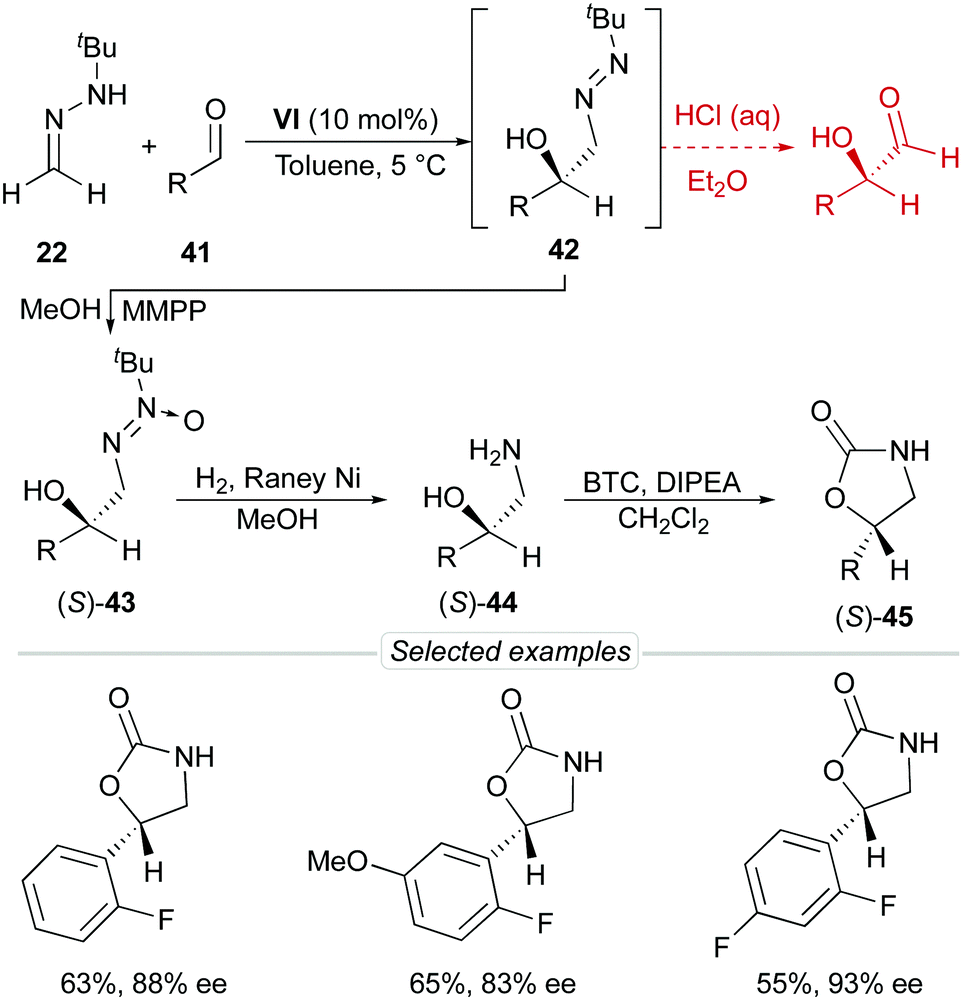
p-Chlorobenzaldehyde 41a, a relative reactive aromatic aldehyde, was chosen as model substrate for preliminary experiments. The thermal reaction between 22 and 41a, in toluene at room temperature, proved to be reversible. Therefore, diazene 42a was transformed in situ into its azoxy compound 43a which could be isolated and analyzed by chromatographic techniques. BINAM bis-(thio)urea catalysts I or II failed in this particular case, affording products in racemic way. Preliminary screenings employing many bifunctional thioureas with different architectures evidenced the need of more active (acidic) catalysts. Thus, an additional screening was performed, leading to the identification of bifunctional squaramides derived from L-tert-leucine as the most promising structures. Initially, moderate levels of asymmetric induction (up to 50% ee) were reached employing VI, in toluene at 5 °C (Scheme 13). The solubility of VI in toluene (<3 mg mL−1 at room temperature) was considered as a possible explanation. This circumstance is frequently associated to the formation of self-aggregates through hydrogen bonds into head-to-tail ladder networks,38 a fact that could be confirmed by X-ray diffraction analysis as shown in Fig. 4, and that frequently limits further implementation of squaramides in asymmetric organocatalysis. To our delight, though, the higher solubility of VI in α,α,α-trifluorotoluene (∼16 mg mL−1 at room temperature) resulted in homogeneous reaction media in which higher conversions (95%) and enantioselectivities (up to 76% ee) were obtained with reliable reproducibility. Interestingly, comparison of the catalytic performance of VI with those of related squaramides bearing different H-bond donor groups, such as pentafluoroaniline (VII) and [3,5-bis(trifluoromethyl)benzyl]amine (VIII) derivatives allows to establish a direct correlation between the H-bond donor ability of the most acidic NH moiety of the catalysts [VI > VII > VIII (inferred from chemical shift of NH protons in 1H NMR)] and the observed enantioselectivities, which drop to 55% and 23% ee for less acidic VII and VIII, respectively (Fig. 5). Temperature and concentration were further optimized: performing the reaction with catalyst VI at −10 °C (0.5 M), followed by in situ N-oxidation, afforded 43a in 90% yield and 90% ee.
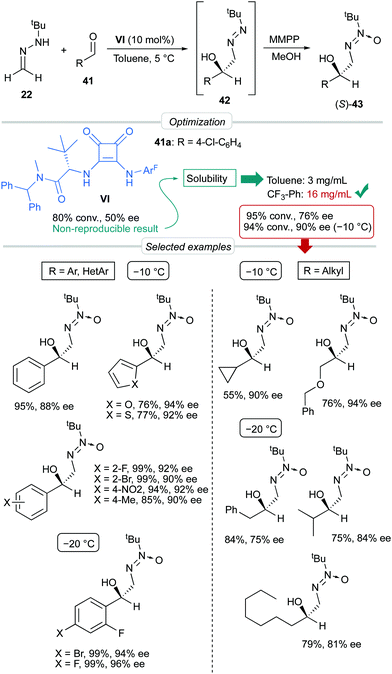 |
| | Scheme 13 Asymmetric addition of FTBH to simple aldehydes. | |
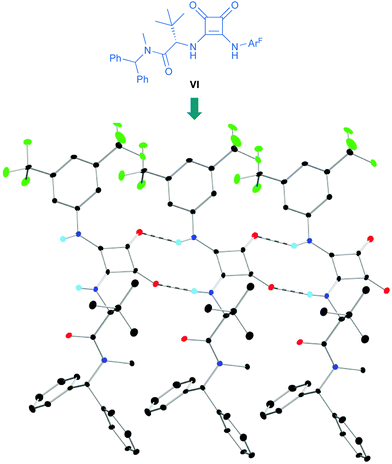 |
| | Fig. 4 Head-to-tail hydrogen-bonding interactions in the solid state of VI (three monomers shown). | |
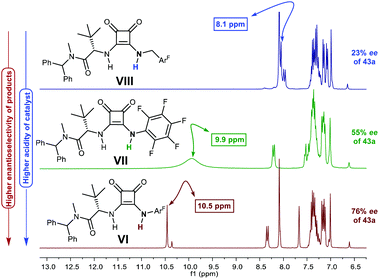 |
| | Fig. 5 Correlation of catalysts’ H-bond donor ability with enantioselectivity of 43a (1H NMR of aromatic region: 6.5–13.0 ppm shown). | |
The scope of the reaction was explored employing different aromatic and heteroaromatic aldehydes; the corresponding azoxy compounds 43 were isolated in excellent yields and high enantioselectivities (up to 96% ee). Remarkably, aliphatic aldehydes with representative types of alkyl chains (cyclopropyl, benzyloxymethyl, benzyl, isopropyl, and n-heptyl) were also tolerated.
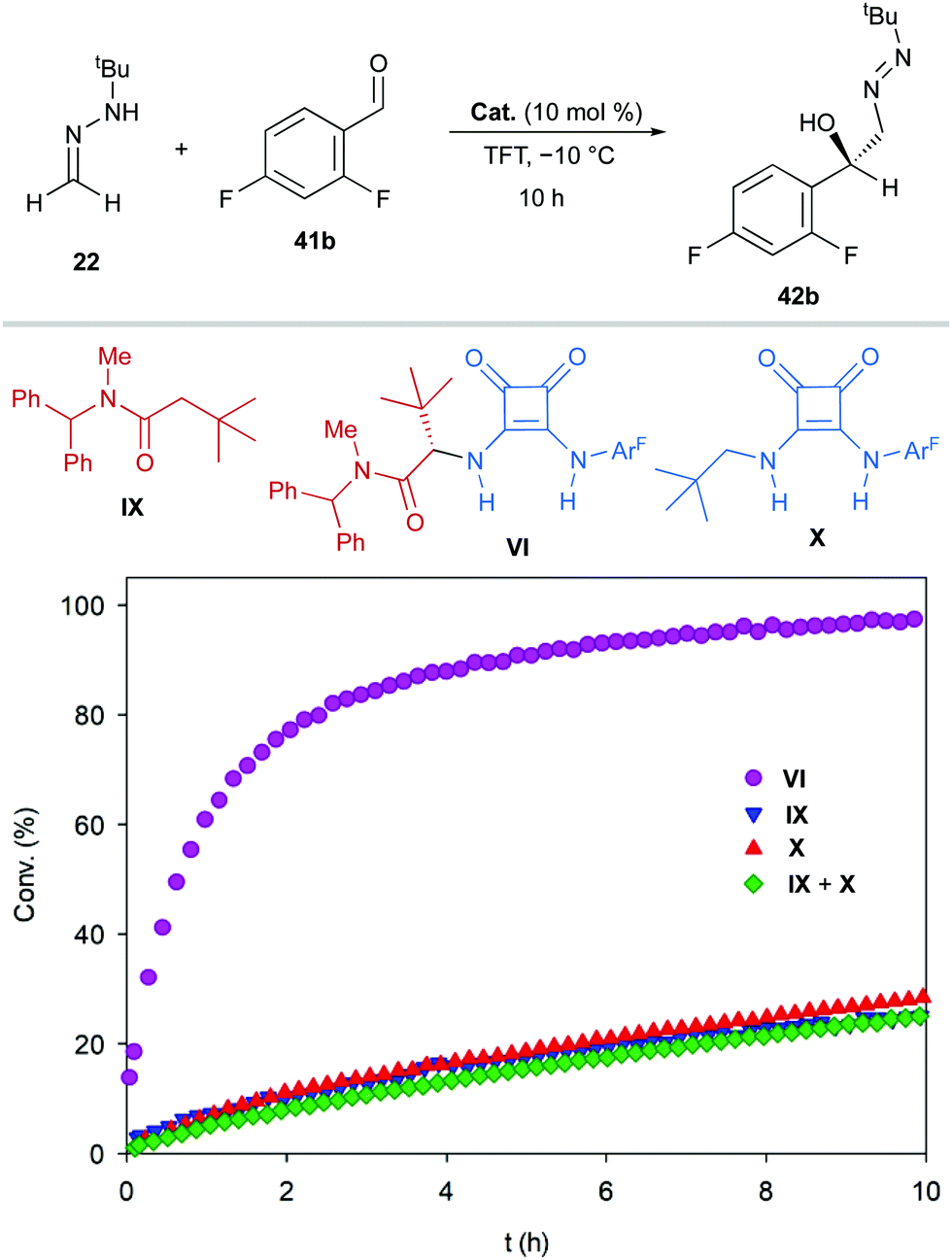
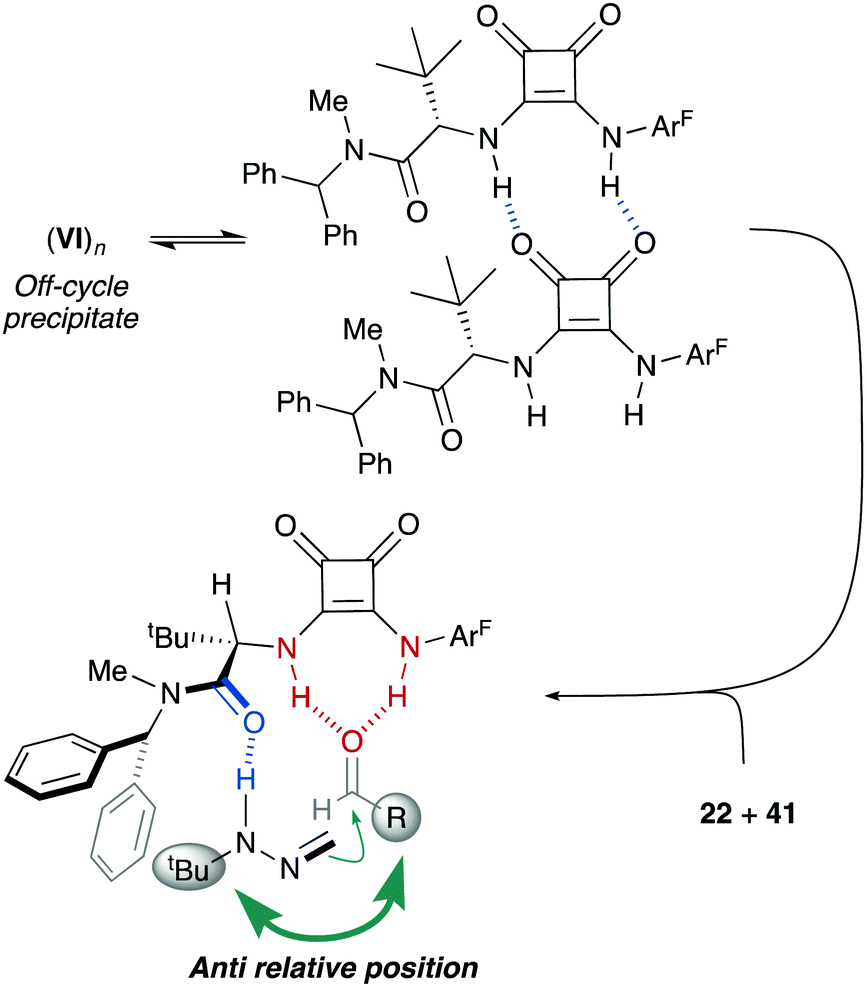
Experimental support for the bifunctional mode of action by squaramide VI was obtained from experiments carried out with monofunctional achiral catalysts IX and X. Thus, these catalysts were evaluated in the reaction between FTBH 22 and 2,6-difluorobenzaldehyde 41b in α,α,α-trifluorotoluene at −10 °C (Scheme 14). The evolution of these control reactions over time (monitored by 19F NMR) showed a much higher catalytic activity of VI (>90% conv. after 10 h) compared to those of IX and X, either acting individually or combined (<30% conv.)39 Consequently, a stereochemical model based on a dual activation of both reagents was proposed, accounting for the observed high enantioselectivities and absolute configuration (Fig. 6). The aldehyde is believed to be activated by the squaramide moiety through H-bonding, while the amide carbonyl group behaves as an efficient H-bond acceptor for the hydrazone NH donor. The activation and the positioning of both reagents drive the preferential approach of azomethine carbon to the Re face of the carbonyl group, minimizing steric repulsions during the C–C bond formation. A slightly negative nonlinear effect suggested a more complex scenario involving insoluble off-cycle catalyst homochiral self-aggregates.40 However, the observed effect was relatively low as reactions become completely homogeneous upon mixing of reagents.
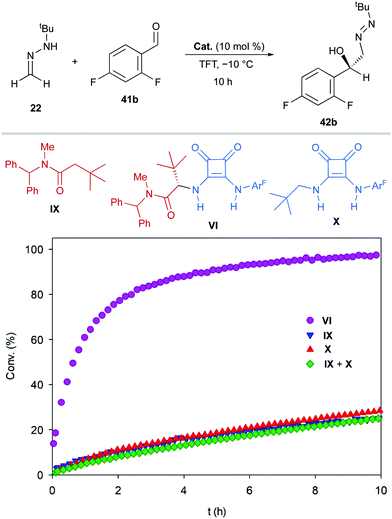 |
| | Scheme 14 Evolution over time for the addition of FTBH (22) to 2,6-difluorobenzaldehyde (41b) in the presence of bifunctional catalyst VI and monofunctional catalysts IX and/or X. | |
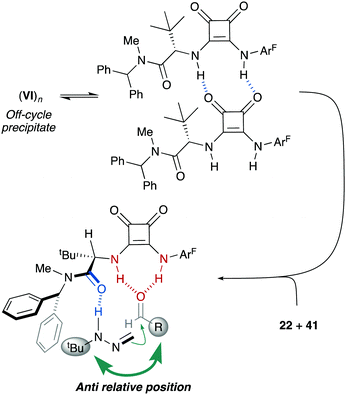 |
| | Fig. 6 Off-cycle catalyst homochiral self-aggregates and stereochemical model. | |
Despite all efforts, the inherent instability of diazenes 42 made it impossible to obtain α-hydroxy aldehydes in this particular case. Alternatively, azoxy compounds 43 were transformed into free amino alcohols 44 by applying standard hydrogenation conditions [RANEY® Ni, H2 (25–50 bar), room temperature]. This unprecedented transformation involves three consecutive steps [hydrogenolytic N–O bond cleavage (azoxy-to-azo compound), NN bond hydrogenation (azo-to-hydrazine reduction) and N–N bond hydrogenolysis (hydrazine-to-amine)] and afforded amino alcohols 44 and derivatives such as oxazolidinones 4541 in good overall yields and without significant erosion of enantioselectivity (Scheme 15).
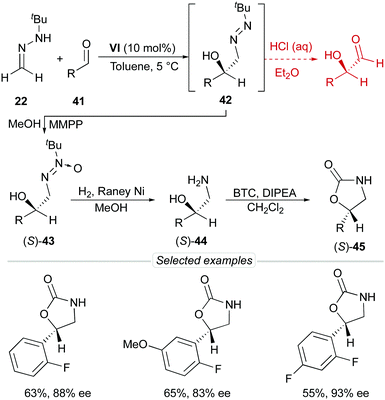 |
| | Scheme 15 Synthesis of amino alcohols 44 and oxazolidinones 45. | |
Di/tri-fluoromethylketones.
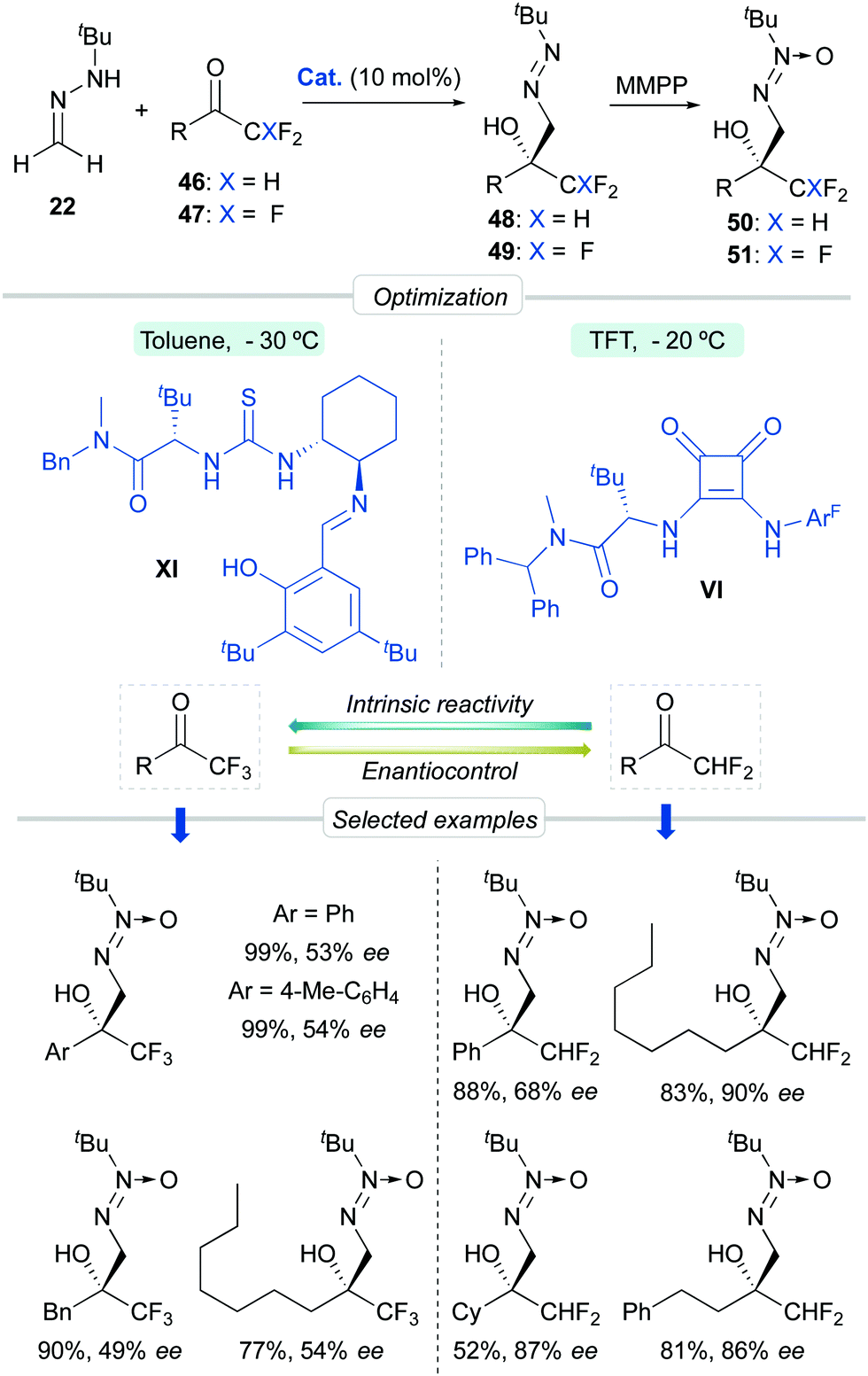
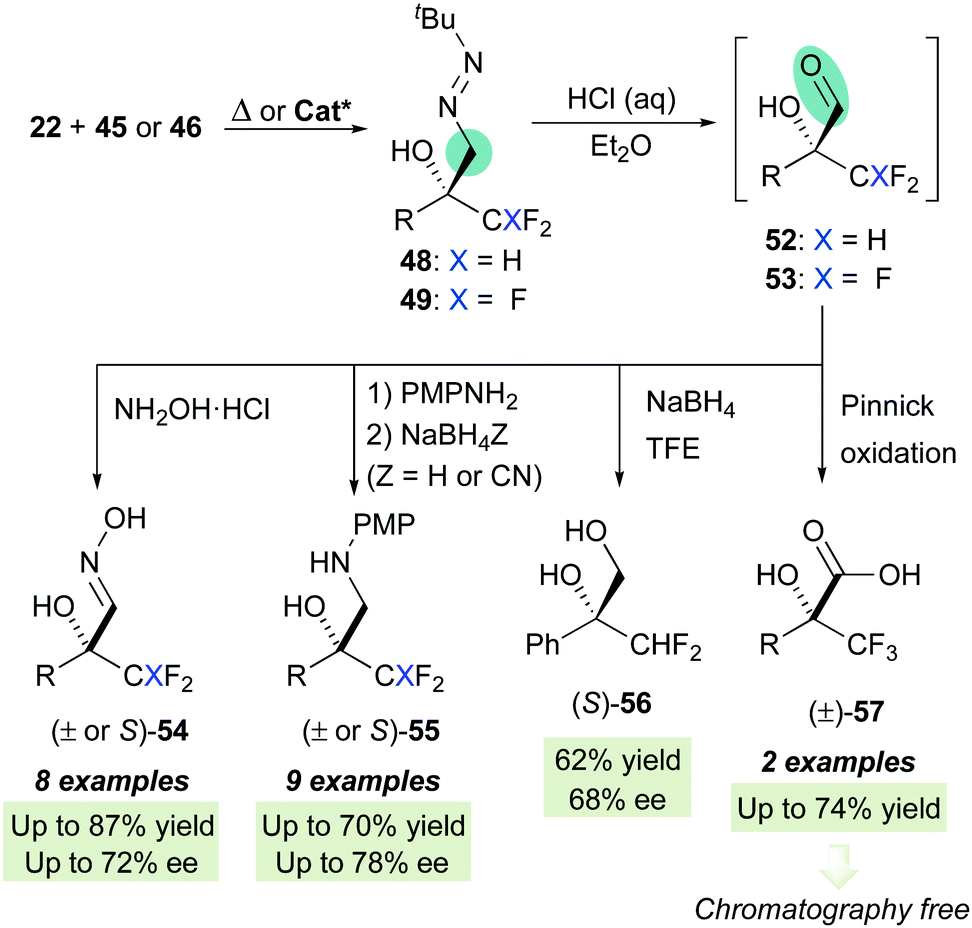
Aiming to further expand the scope of this catalytic system, we next focused on the asymmetric functionalization of di- and tri-fluoromethylketones 46 and 47 for the synthesis of enantioenriched, densely functionalized fluorinated alcohols 48 and 49, respectively (Scheme 16). With respect to the previous cases, this can be considered a more challenging task because of two major predictable difficulties: (a) the uncatalyzed background reaction is enhanced by the strong inductive effect of the CHF2/CF3 groups and (b) The sizes of the CHF2 or CF3 groups and the other ketone substituent are not so different, making more difficult for the catalyst to discriminate geometries based in steric differentiation. The optimization processes, however, showed that L-tert-leucine-derived H-bonding organocatalysts offer again satisfactory enantioselectivities. For difluoromethylketones, squaramide VI (in trifluorotoluene at −20 °C) provided high activities and moderate-to-good enantioselectivities, while for trifluoromethylketones, multifunctional thiourea XI (in toluene at −30 °C) was a slightly better catalyst, albeit enantiocontrol was in both cases hampered, as anticipated, by fast, unavoidable background reactions. In parallel, the high reactivity of trifluoromethylketones was exploited for the development of a green nucleophilic formylation strategy based on a solvent-free reaction/hydrolysis sequence.42 Diazenes 48–49 were subsequently transformed in a ‘one-pot’ fashion into α-hydroxy α-trifluoro(difluoro)methyl aldehydes 52–53via a tautomerization-hydrolysis sequence. These crude aldehydes do not tolerate chromatographic purification but were isolated with a high degree of purity (>95%), thereby validating the whole formylation methodology. Subsequent derivatizations provide synthetically useful fluorinated intermediates such as oximes 54, β-aminoalcohols 55, diol 56 and α-hydroxy acids 57 (Scheme 17).
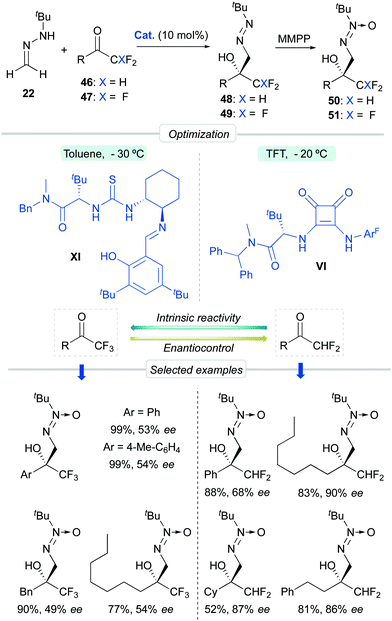 |
| | Scheme 16 Asymmetric addition of FTBH to fluorinated methylketones. | |
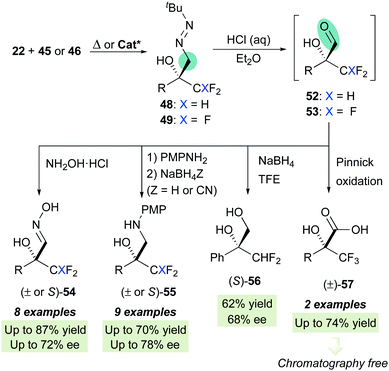 |
| | Scheme 17 Unmasking by hydrolysis and synthetic sequence to synthetically relevant fluorinated targets. | |
4. Conclusions
The combination of formaldehyde tert-butyl hydrazone, as a neutral formyl anion equivalent, with bifunctional H-bond donor/acceptor organocatalysts has emerged as a versatile tool for the asymmetric formylation of carbonyl compounds. The design of a dual activation strategy has been key to successfully perform highly enantioselective nucleophilic additions, formally aza-carbonyl-ene reactions, with a broad scope of substrates including α-keto esters, isatines, α-keto phosphonates, simple aldehydes and fluorinated ketones, efficiently yielding tert-butyl azomethyl carbinols as the primary reaction products. In these reactions, ternary catalyst–reagent–substrate complexes are generated, in which the carbonyl compound of the substrate is activated by multiple H-bond interactions (with a urea/thiourea/squaramide moiety), while the nucleophilicity of the hydrazone reagent is enhanced by an additional H-bonding interaction with the basic oxygen atom of an urea or amide group. In most cases, the formylation procedure is efficiently completed after a simple ‘one-pot’ transformation of the obtained diazenes into the targeted aldehydes via tautomerization followed by hydrolytic cleavage of the hydrazone intermediates. Moreover, a variety of other densely functionalized chiral building blocks can be also obtained after simple functional group transformations. Despite the high reactivity observed for some substrates, none of the catalysts used allowed to expand the reactivity to N-tert-butylhydrazones from higher aldehydes.43 Future investigations based in alternative catalysts and/or activation strategies, therefore, will be needed to overcome this limitation and develop a more general method for the asymmetric acylation of carbonyl compounds. Moreover, the implementation of some of these approaches in target-oriented synthesis might be also explored for accessing on-demand biologically active targets.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This work was supported by the Spanish MINECO (CTQ2016-76908-C2-1-P, CTQ2016-76908-C2-2-P), European FEDER funds and the Junta de Andalucía (Grants P18-FR-3531 and US-1262867).
Notes and references
-
(a) D. Seebach, Angew. Chem., Int. Ed. Engl., 1979, 18, 239 CrossRef;
(b)
T. A. Hase, Umpoled Synthons: A survey of Sources and Uses in Synthesis, Wiley Interscience, New York, 1987 Search PubMed.
-
(a) Review: J. Johnson, Angew. Chem., Int. Ed., 2004, 43, 1326 CrossRef CAS PubMed . See also: ;
(b) D. Enders and U. Kallfass, Angew. Chem., Int. Ed., 2002, 41, 1743 CrossRef CAS;
(c) S. E. Denmark and T. W. Wilson, Nat. Chem., 2010, 2, 937 CrossRef CAS PubMed;
(d) D. Enders, A. Grossmann, J. Fronert and G. Raabe, Chem. Commun., 2010, 46, 6282 RSC.
- Reviews:
(a) D. Enders and T. Balensiefer, Acc. Chem. Res., 2004, 37, 534 CrossRef CAS PubMed;
(b) M. Christmann, Angew. Chem., Int. Ed., 2005, 44, 2632 CrossRef CAS PubMed;
(c) J. S. Johnson, Angew. Chem., Int. Ed., 2004, 43, 1326 CrossRef CAS PubMed;
(d) X. Bugaut and F. Glorius, Chem. Soc. Rev., 2012, 41, 3511 RSC.
- T. Matsumoto, H. Yamamoto and S. Inoue, J. Am. Chem. Soc., 1984, 106, 4829 CrossRef CAS.
- A. Kirschning, C. Kujat, S. Luiken and E. Schaumann, Eur. J. Org. Chem., 2007, 2387 CrossRef CAS and references therein.
- L. Colombo, C. Gennari and C. Scolastico, J. Chem. Soc., Chem. Commun., 1979, 591 RSC.
- B. Delouvrié, F. Nájera, L. Fensterbank and M. Malacria, J. Organomet. Chem., 2002, 643–644, 130 CrossRef.
- R. E. Gawley, S. A. Campagna, M. Santiago and T. Ren, Tetrahedron: Asymmetry, 2002, 13, 29 CrossRef CAS.
-
(a) C. Gaul and D. Seebach, Org. Lett., 2000, 2, 1501 CrossRef CAS PubMed;
(b) C. Gaul, K. Schärer and D. Seebach, J. Org. Chem., 2001, 66, 3059 CrossRef CAS PubMed.
- S. Nakamura, Y. Ito, L. Wang and T. Toru, J. Org. Chem., 2004, 69, 1581 CrossRef CAS PubMed.
-
(a) L. Wang, S. Nakamura and T. Toru, Org. Biomol. Chem., 2004, 2, 2168 RSC;
(b) L. Wang, S. Nakamura, Y. Ito and T. Toru, Tetrahedron: Asymmetry, 2004, 15, 3059 CrossRef CAS.
-
(a) J. M. Lassaletta and R. Fernández, Synlett, 2000, 1228 Search PubMed;
(b) R. Brehme, D. Enders, R. Fernández and J. M. Lassaletta, Eur. J. Org. Chem., 2007, 5629 CrossRef CAS . The nucleophilicity of formaldehyde N,N-dialkylhydrazones has been measured and correlated with their ambident reactivity: ;
(c) M. Biplab, T. Konstantin and M. Herbert, Angew. Chem., Int. Ed., 2013, 52, 11900 CrossRef PubMed.
-
D. Monge and R. Fernández, (S)-2-Methoxymethyl-1-methylideneamino-pyrrolidine, Encyclopedia of Reagents for Organic Synthesis, John Wiley and Sons, 2019 Search PubMed.
-
(a) J. M. Lassaletta, R. Fernández, E. Martín-Zamora and C. Pareja, Tetrahedron Lett., 1996, 37, 5787 CrossRef CAS;
(b) R. Fernández, E. Martín-Zamora, C. Pareja and J. M. Lassaletta, J. Org. Chem., 2001, 66, 5201 CrossRef PubMed.
- R. Fernández, E. Martín-Zamora, C. Pareja, M. Alcarazo, J. Martín and J. M. Lassaletta, Synlett, 2001, 1158 CrossRef.
-
(a) R. Fernández, E. Martín-Zamora, C. Pareja, J. Vázquez, E. Díez, A. Monge and J. M. Lassaletta, Angew. Chem., Int. Ed., 1998, 37, 3428 CrossRef;
(b) C. Pareja, E. Martín-Zamora, R. Fernández and J. M. Lassaletta, J. Org. Chem., 1999, 64, 8846 CrossRef CAS PubMed.
- D. Enders, L. Wortmann and R. Peters, Acc. Chem. Res., 2000, 33, 157 CrossRef CAS PubMed.
- R. Fernández, C. Gasch, J. M. Lassaletta, J. M. Llera and J. Vázquez, Tetrahedron Lett., 1993, 34, 141 CrossRef.
- Some exceptions:
(a) D. Monge, E. Martín-Zamora, J. Vázquez, M. Alcarazo, E. Álvarez, R. Fernández and J. M. Lassaletta, Org. Lett., 2007, 9, 2867 CrossRef CAS PubMed;
(b) S. Breitler and E. M. Carreira, J. Am. Chem. Soc., 2015, 137, 5296 CrossRef CAS PubMed.
- Hydrazones in asymmetric organocatalysis: M. G. Retamosa, E. Matador, D. Monge, J. M. Lassaletta and R. Fernández, Chem. – Eur. J., 2016, 22, 13430 CrossRef CAS PubMed.
-
(a) R. P. Herrera, D. Monge, E. Martín-Zamora, R. Fernández and J. M. Lassaletta, Org. Lett., 2007, 9, 3303 CrossRef CAS PubMed . For selected previous diastereoselective Michael additions of chiral formaldehyde N,N-dialkylhydrazones, see: ;
(b) J. M. Lassaletta, R. Fernández, E. Martín-Zamora and E. Díez, J. Am. Chem. Soc., 1996, 118, 7002 CrossRef CAS;
(c) J. Vázquez, A. Prieto, R. Fernández, D. Enders and J. M. Lassaletta, Chem. Commun., 2002, 498 RSC;
(d) J. Vázquez, E. Cristea, E. Díez, J. M. Lassaletta, A. Prieto and R. Fernández, Tetrahedron, 2005, 61, 4115 CrossRef.
-
(a) D. J. Dixon and A. L. Tillman, Synlett, 2005, 2635 CrossRef CAS;
(b) M. Rueping, E. Sugiono, T. Theissmann, A. Kuenkel, A. Köckritz, A. Pews-Davtyan, N. Nemati and M. Beller, Org. Lett., 2007, 9, 1065 CrossRef CAS PubMed . For examples of 1,2-additions of hydrazones from higher aldehydes, see: ;
(c) T. Hashimoto, M. Hirose and K. Maruoka, J. Am. Chem. Soc., 2008, 130, 7556 CrossRef CAS PubMed;
(d) T. Hashimoto, H. Kimura and K. Maruoka, Angew. Chem., Int. Ed., 2010, 49, 6844 CrossRef CAS PubMed.
- J. E. Baldwin, R. M. Adlington, J. C. Bottaro, J. N. Kolhe, M. W. D. Perry and A. U. Jain, Tetrahedron, 1986, 42, 4223 CrossRef CAS.
- J. E. Baldwin, R. M. Adlington, A. U. Jain, J. N. Kolhe and M. W. D. Perry, Tetrahedron, 1986, 42, 4247 CrossRef CAS.
-
(a) A. M. Crespo-Peña, D. Monge, E. Martín-Zamora, E. Álvarez, R. Fernández and J. M. Lassaletta, J. Am. Chem. Soc., 2012, 134, 12912 CrossRef PubMed;
(b) J. A. Carmona, G. de Gonzalo, I. Serrano, A. M. Crespo-Peña, M. Šimek, D. Monge, R. Fernández and J. M. Lassaletta, Org. Biomol. Chem., 2017, 15, 2993 RSC.
- D. Monge, A. M. Crespo-Peña, E. Martín-Zamora, E. Álvarez, R. Fernández and J. M. Lassaletta, Chem. – Eur. J., 2013, 19, 8421 CrossRef CAS PubMed.
- I. Serrano, D. Monge, E. Álvarez, R. Fernández and J. M. Lassaletta, Chem. Commun., 2015, 51, 4077 RSC.
- For a related study showing the participation of the ortho CH bonds of the 3,5-bis(trifluoromethyl)phenyl group as donors H-bond networks, see: Z. Zhang, K. M. Lippert, H. Hausmann, M. Kotke and P. R. Schreiner, J. Org. Chem., 2011, 76, 9764 CrossRef CAS PubMed.
- For a closely related structure in solid state, see: R. Holakovský, M. Pojarová, M. Dusek, J. Cejka and I. Císarová, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2011, 67, o384 CrossRef PubMed.
- K. Tamura, M. Furutachi, N. Kumagai and M. Shibasaki, J. Org. Chem., 2013, 78, 11396 CrossRef CAS PubMed and references therein.
- H. K. Munayyer, P. A. Mann, A. S. Chau, T. Yarosh-Tomaine, J. R. Greene, R. S. Hare, L. Heimark, R. E. Palermo, D. Loebenberg and P. M. McNicholas, Antimicrob. Agents Chemother., 2004, 48, 3690 CrossRef CAS PubMed.
-
(a) I. Ojima, C. M. Sun, M. Zucco, Y. H. Park, O. Duclo and S. Kuduk, Tetrahedron Lett., 1993, 34, 4149 CrossRef CAS;
(b) I. Ojima, T. Wang and F. Delaloge, Tetrahedron Lett., 1998, 39, 3663 CrossRef CAS;
(c) G. Cardillo, L. Gentilucci, A. Tolomelli and C. Tomasini, J. Org. Chem., 1998, 63, 2351 CrossRef CAS;
(d) Y. Takeda, T. Yoshino, K. Uoto, J. Chiba, T. Ishiyama, M. Iwahana, T. Jimbo, N. Tanaka, H. Terasawa and T. Soga, Bioorg. Med. Chem. Lett., 2003, 13, 185 CrossRef CAS PubMed.
- S. Peddibhotla, Curr. Bioact. Compd., 2009, 5, 20 CrossRef CAS.
- Selected examples of bioactive azoxy compounds:
(a) The antimicrobial and cytotoxic elaiomycins: L. Ding, B. L. T. Ndejouong, A. Maier, H. H. Fiebig and C. Hertweck, J. Nat. Prod., 2012, 75, 1729 CrossRef CAS PubMed;
(b) The antibiotic valanimycin: R. P. Garg, X. L. L. Qian, L. B. Alemany, S. Moran and R. J. Parry, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 6543 CrossRef CAS PubMed;
(c) Novel antifungal antibiotics maniwamycins A and B: M. Nakayama, Y. Takahashi, H. Itoh, K. Kamiya, M. Shiratsuchi and G. Otani, J. Antibiot., 1989, 42, 1535 CrossRef CAS PubMed.
-
(a) For a review on naturally occurring β-amino-α-hydroxyphosphonic acid derivatives, see: P. Mastałerz and P. Kafarski, in Aminophosphonic and Aminophosphinic Acids-Chemistry and Biological Activity, ed. V. P. Kukhar and H. R. Hudson, John Wiley, Chichester, 2000, pp 1–31 Search PubMed;
(b) D. V. Patel, K. Rielly-Gauvin, D. E. Ryono, C. A. Free, W. L. Rogers, S. A. Smith, J. M. DeForrest, R. S. Oehl and E. W. Petrillo, J. Med. Chem., 1995, 38, 4557 CrossRef CAS PubMed.
- E. Matador, M. G. Retamosa, D. Monge, J. Iglesias-Sigüenza, R. Fernández and J. M. Lassaletta, Chem. – Eur. J., 2018, 24, 6854 CrossRef CAS PubMed.
- E. Matador, M. G. Retamosa, A. Jiménez-Sánchez, D. Monge, R. Fernández and J. M. Lassaletta, Eur. J. Org. Chem., 2019, 130 CrossRef CAS.
- See:
(a) A. Portell, R. Barbas, D. Braga, M. Polito, C. Puigjanerand and R. Prohens, CrystEngComm, 2009, 11, 52 RSC;
(b) A. Portell, M. Font-Bardia, A. Bauzá, A. Frontera and R. Prohens, Cryst. Growth Des., 2014, 14, 2578 CrossRef;
(c) A. Portell, M. Bardia-Font, A. Bauzá, A. Frontera and R. Prohens, CrystEngComm, 2016, 18, 6437 RSC.
- For a discussion on multiple vs multifunctional catalysis, see: S. Piovesana, D. M. S. Schietroma and M. Bella, Angew. Chem., Int. Ed., 2011, 50, 6216 CrossRef CAS PubMed.
- In contrast, Jacobsen and co-workers described a positive non-linear effect due to the presence of soluble off-cycle catalysts homochiral self-aggregates for related amide-thioureas in different contexts. See:
(a) D. D. Ford, D. Lehnherr, C. R. Kennedy and E. N. Jacobsen, J. Am. Chem. Soc., 2016, 138, 7860 CrossRef CAS PubMed;
(b) D. Ford, D. Lehnherr, C. R. Kennedy and E. N. Jacobsen, ACS Catal., 2016, 6, 4616 CrossRef CAS PubMed.
- Oxazolidinones in medicinal chemistry:
(a) T. A. Mukhtar and G. D. Wright, Chem. Rev., 2005, 105, 529 CrossRef CAS PubMed;
(b) M. R. Barbachyn and C. W. Ford, Angew. Chem., Int. Ed., 2003, 42, 2010 CrossRef CAS PubMed;
(c) H. Kakeya, M. Morishita, K. Kobinata, M. Osono, M. Ishizuka and J. Osada, J. Antibiot., 1998, 51, 1126 CrossRef CAS PubMed;
(d) M. F. Gordeev and Y. Y. Zhengyu, J. Med. Chem., 2014, 57, 4487 CrossRef CAS PubMed.
- E. Matador, D. Monge, R. Fernández and J. M. Lassaletta, Green Chem., 2016, 18, 4042 RSC.
- Recent example on the asymmetric 1,2-addition of N-tert-butylhydrazones to imines employing Brønsted-acid catalysis: Y. Wang, Q. Wang and J. Zhu, Angew. Chem., Int. Ed., 2017, 56, 5612 CrossRef CAS PubMed.
Footnotes |
| † Dedicated to the memory of Professor Kilian Muñiz. |
| ‡ These authors contributed equally to this work. |
|
| This journal is © The Royal Society of Chemistry 2020 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence a,
María
de Gracia Retamosa‡
a,
María
de Gracia Retamosa‡