
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Manganese-catalysed transfer hydrogenation of esters†‡
Conor L.
Oates
,
Magnus B.
Widegren
and
Matthew L.
Clarke
 *
*
School of Chemistry, University of St Andrews, EaStCHEM, St Andrews, Fife, UK. E-mail: mc28@st-andrews.ac.uk; Fax: +44 (0)1334 463808; Tel: +44 (0)1334 463850
First published on 11th June 2020
Abstract
Manganese catalysed ester reduction using ethanol as a hydrogen transfer agent in place of dihydrogen is reported. High yields can be achieved for a range of substrates using 1 mol% of a Mn(I) catalyst, with an alkoxide promoter. The catalyst is derived from a tridentate P,N,N ligand.
The hydrogenation of esters has changed from being a very problematic aspirational transformation 15 years ago, to a highly efficient example of homogeneous catalysis. There are quite a few effective Ru-based catalysts,1 but also Mn and Fe based systems that show promise.2,3 The use of molecular hydrogenation is frequently the cleanest and most economic reductant, but there are a variety of scenarios where the use of a liquid reductant may be desirable. The importance of transfer hydrogenation, using an alcohol as hydrogen donor, to catalytic reductions cannot be doubted; there are many useful practical applications for asymmetric transfer hydrogenation of ketones.4a–f
Surprisingly, given the level of interest in transfer hydrogenation, relatively few papers have described transfer hydrogenation of esters (Fig. 1).5a–d Dubey and Khaskin reported the use of just 1 mol% of a ruthenium catalyst, using ethanol as reductant and an alkoxide co-catalyst.5b The discovery by de Vries and co-workers of an effective base-free Fe borohydride catalyst using ethanol as reductant, this time requiring 5 mol% catalyst loading, is a significant milestone.5d Catalysts such as this, which are derived from an indefinitely sustainable metal source, and that permit higher catalyst impurities than the 5–10 ppm generally required as a minimum for precious metal based catalysts6 are highly desirable. This significance has spurred a lot of recent research on Mn catalysed reductions.
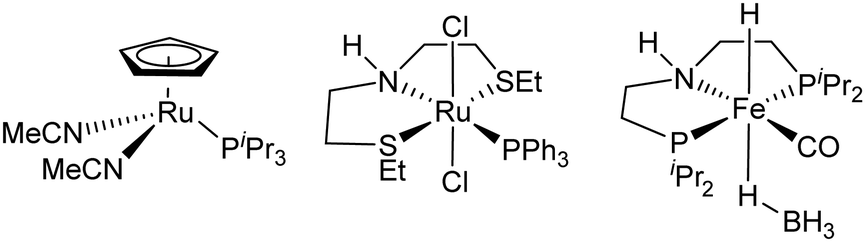 | ||
| Fig. 1 Catalysts previously used for transfer hydrogenation of esters.5 | ||
We have recently reported the use of catalyst 1 (Fig. 2) (in racemic form) in some synthetically useful ester hydrogenations using hydrogen as reductant,3 and more recently rationally designed second generation systems, 2 and 3 (in enantiomerically pure form) for highly efficient enantioselective hydrogenation of a broad range of ketones (0.01 to 0.1 mol% catalyst loadings).7 These are amongst the most active of all non-precious metal catalysts for the hydrogenation of esters and ketones. That being said, transfer hydrogenation of ketones using any of these catalysts was a more mixed picture, with only a handful of typical substrates giving good results.7 De Vries and co-workers also found that a Mn/PNP hydrogenation catalyst was entirely ineffective for ester transfer hydrogenation.5d To our surprise, given this background, we have now found that a highly effective transfer hydrogenation of a range of esters is possible using the Mn catalysts 1 or 2, and report this discovery here.
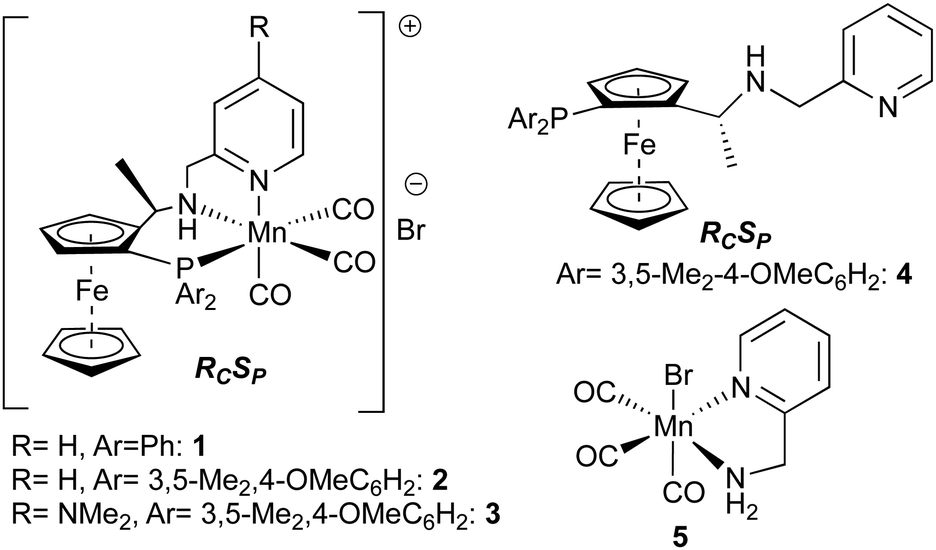 | ||
| Fig. 2 Catalysts investigated in this work. | ||
Our objective was to enable a Mn-based catalyst to conduct ester transfer hydrogenation at a catalyst loading below the 5 mol% level used by de Vries and co-workers.5d A selection of our Mn catalysts were investigated using conditions relatively similar to those used in ref. 5b. We chose the reduction of ethyl 4-fluorobenzoate in EtOH as a model substrate to identify suitable conditions (summarised in Table 1). The optimal conditions identified during this study are shown in entry 1. We first identified the most suitable catalysts from those we have recently developed.2d,3,7
|
|
|||
|---|---|---|---|
| Entry | Change from optimal conditions | Conversion of 6c (%) | 7 (%) |
| a Standard reaction conditions: 6 (0.5 mmol), 1-methylnaphthalene (0.15 mmol), base and Mn cat. (0.005 mmol) in dry solvent under N2 atmosphere at 100 °C for 22 h. b 4 (0.05 mmol) and Mn(CO)5Br (0.05 mmol) in dry EtOH at 100 °C for 2 h, then 6 (0.5 mmol), 1-methylnaphthalene (0.15 mmol) and KOtBu (0.10 mmol) added and stirred at 100 °C for 24 h; all under N2 atmosphere. c Determined by 1H NMR using 1-methylnaphthalene as internal standard. Isolated yields (after chromatographic purification) in brackets. | |||
| 1 | — | 90 | 76 (74) |
| 2 | 1 mol% 1 | 85 | 74 |
| 3 | 1 mol% 3 | 76 | 57 |
| 4 | (1 mol% 4 + [Mn(CO)5Br])b | 90 | 76 (73) |
| 5 | 1 mol% 5 | 15 | 2 |
| 6 | No catalyst | 13 | Trace |
| 7 | 10 mol% KOtBu | 81 | 67 |
| 8 | 30 mol% KOtBu | 91 | 52 |
| 9 | 0.25 M | 46 | 43 |
| 10 | 0.5 M | 66 | 53 |
| 11 | 1 M | 84 | 76 |
| 12 | 20 mol% K2CO3 | 84 | 67 |
| 13 | MeOH as solvent | 93 | 19 |
| 14 | n BuOH as solvent | 19 | Trace |

The use of either 1 mol% catalyst 1 or 2 gave similarly good yields. Use of catalyst 3 (entry 3) led to poorer conversions. This latter catalyst is slightly more active in pressure-hydrogenation of esters below around 70 °C.7 The kinetic studies described in ref. 7 show the catalyst degrades during the reaction above this temperature; the lower yield here is likely due to its lower thermal stability. The optimal base loading was found to be 20 mol% of KOtBu: lowering to 10 mol% (entry 7) led to decreased conversion of starting material and raising to 30 mol% (entry 8) lead to lower product formation. Pleasingly, changing to the weaker K2CO3 at 20 mol% (entry 12) still produced useful levels of product albeit with lower yield. We found the optimal concentration (wrt ester substrate) to be 2 M. Lower concentrations (entries 9/10) resulted in lower conversions to (7). The alternative primary alcohol hydrogen donors we have examined are less effective than EtOH (entries 13 and 14). Using MeOH (2 M) resulted in mostly transesterification to the methyl ester and only a small amount of product formation. Use of nBuOH (2 M) lead to almost no product being formed. Therefore, optimal conditions for the reduction of 6 were 1 mol% of 2 in EtOH with a 2 M concentration at 100 °C for 22 h.§
While the use of well-defined pre-catalysts has many advantages, sometimes it is preferable to generate a catalyst in situ. Pre-stirring 1 mol% of both 4 and [Mn(CO)5Br] for 2 h at 100 °C in EtOH and then adding 6/base before stirring for a further 24 h at 100 °C (entry 4, Table 1) produced a comparable yield of (7) as when well-defined 2 was used in the standard conditions (we note that the yield drops to 63% if there is no pre-stirring period for complexation, not shown).
With optimal conditions in hand we began investigating the reaction scope (with the results summarised in Table 2); at first focussing on aromatic ethyl esters to eliminate transesterification between solvent and substrate. Further demonstration of halogen tolerance was shown when ethyl 4-iodobenzoate (8a) was reduced in good yield, as were unfunctionalised esters, (8b) (8c). Examples of both oxygen and nitrogen containing heterocycles are tolerated by the conditions-demonstrated by the reduction of ethyl 2-furoate (8d) and ethyl isonicotinate (8e) in good yields. While ether functionality, even in the ortho position to the ester is unproblematic (8f, 8h), the presence of free amino, hydroxy and nitrile function was problematic thus far.
| a Standard reaction conditions: ester substrate (0.5 mmol), tBuOK (0.1 mmol) and 2 (0.005 mmol) in dry EtOH (0.25 mL) under N2 atmosphere at 100 °C for 22 h. Isolated yields (after chromatographic purification) in brackets. b Determined by 1H NMR using 1,4-dimethoxybenzene as internal standard. c 0.01 mmol of 2. d 0.0075 mmol of 2. |
|---|

|
We were then interested to see if the conditions were applicable to aliphatic ethyl esters; all the examples tested could be reduced with good yields. Reduction of unfunctionalised 8l, 8m, and 8n worked well. The reduction of lauryl and oleyl esters is a process important to the fine chemical industry with oleyl (9o) and lauryl alcohols (9p) seeing wide industrial and commercial use.9 Satisfyingly, the reduction of ethyl laurate (8o) and ethyl oleate (8p) produced their respective alcohols in good yield. (S)-Naproxen (8q) could also be used to produce Naproxol (9q) in very good yield but with epimerisation of the chiral centre. The amino acid ester tolerance of the conditions was investigated by reducing ethyl N,N-dibenzylglycine (8r) generating N,N-dibenzylamino ethanol (9r) in good yield.
Esters derived from alcohols other than ethanol were then investigated. The halogenated methyl 4-chlorobenzoate (8s) and methyl 4-bromobenzoate (8t) were both reduced in very good yield (Table 2). Heterocyclic methyl nicotinate (8u) was reduced too in excellent yield. The α-β-unsaturated ester methyl cinnamate (8v) was totally reduced to the saturated compound 3-phenylpropanol (9m) in good yield. Finally, the methyl (8w) and isopropyl (8x) 2-methoxybenzoate esters produced 2-methoxybenzyl alcohol (9f) in good yields when submitted to the standard conditions. Transesterification was proven to occur using substrate 8x and 8t (see Scheme 2 and ESI†). The transesterification with ethanol occurring under the reaction conditions led us to consider a one-pot esterification–reduction of carboxylic acids. Carboxylic acids cannot be reduced by any of the base-activated Ru or Mn amino-phosphine catalysts, and are very difficult to hydrogenate, despite some notable contributions.10a–d
To prove the viability of this concept we have examined just two randomly selected substrates: the ethyl esters were generated by modified Steglich esterification conditions11 using N,N′-diisopropylcarbodiimide (DIC) and Dimethylaminopyridine (DMAP) in EtOH. Adding catalyst 2 and base and heating then allowed the transfer hydrogenation to proceed with a fairly similar yield to using pure ethyl esters to afford the alcohol (Scheme 1). Since both of these protocols worked similarly to using ethyl esters, we would therefore suggest the catalysts have tolerance to these esterification conditions and esters should be able to be made in situ.
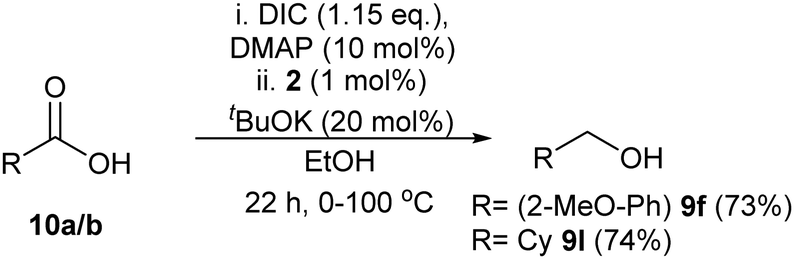 | ||
| Scheme 1 One-pot esterification/reduction of carboxylic acids. For experimental procedure, see ESI.† Isolated yields (after chromatographic purification) in brackets. | ||
When the reaction mixtures are analysed before the reaction completion, very little starting ester remains with various transesterification processes occuring. Using isopropyl (8x) 2-methoxybenzoate ester, there is rapid transesterification with ethanol (see ESI†). The ester formed from the product alcohol, 9t can also be detected in the mixture, as can small amounts of the acetate of the product (Scheme 2). Transfer hydrogenation is an equilibrium process and the conditions used enable high conversion to the desired primary alcohol (and useful amounts isolated in pure form), but complete conversion to the product is not observed, unlike the pressure hydrogenation using hydrogen gas.7 The mechanism is consistent with that discussed in previous papers5 where dehydrogenation of the ethanol reductant, which is in excess occurs alongside ester reduction with the hemi-acetals produced also being dehydrogenated (EtCHOH(OR)) and reduced respectively (Scheme 2). The formation of a Mn-hydride from 1–3 using basic alcohol solutions has resisted our attempts at isolation and even detection: in contrast to similar complexes with Ru and Ir, free ligand is detected if the catalysts are activated without any substrate to reduce. However, based on precedents from a plethora of examples using a variety of metals, this should initiate the cycle.
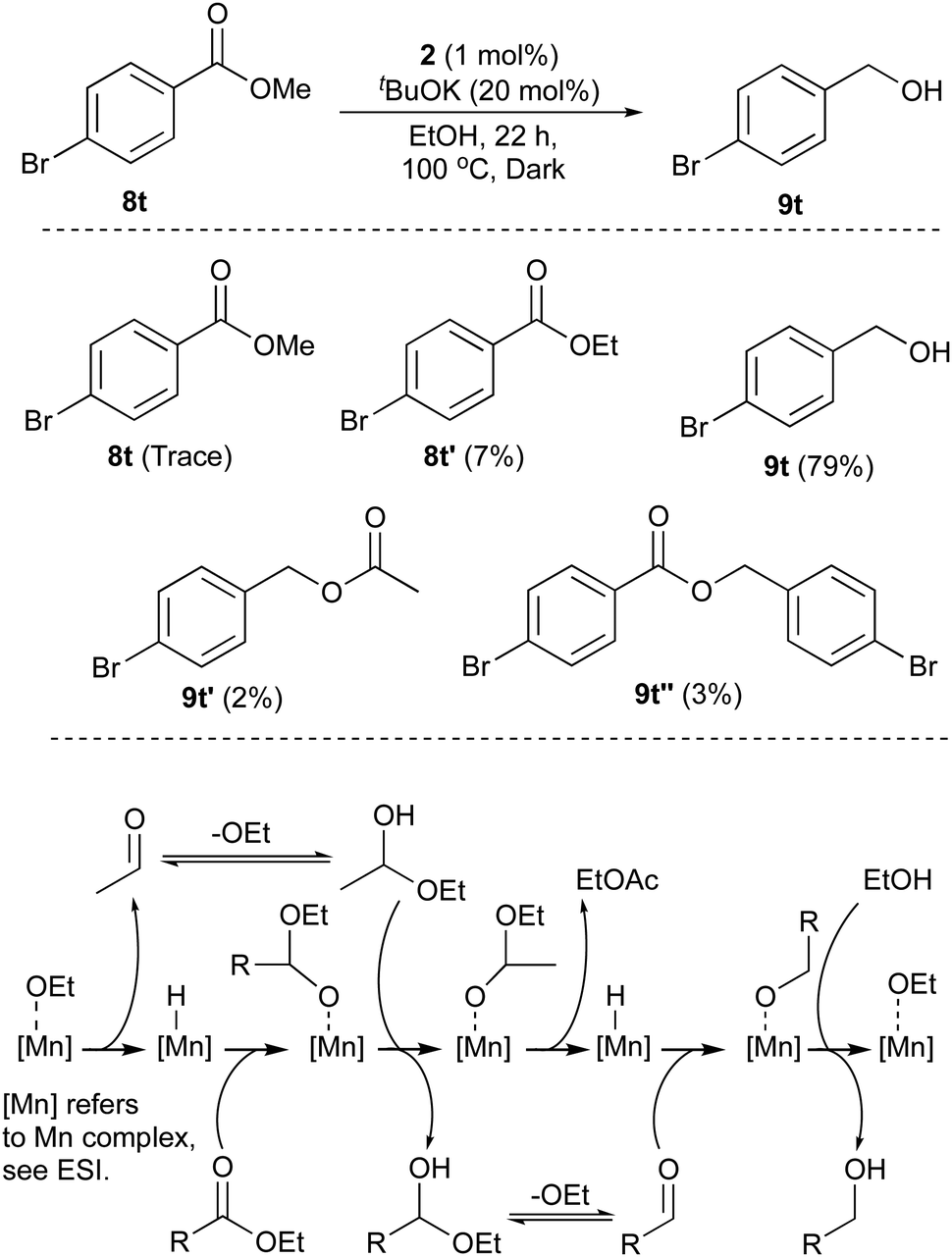 | ||
| Scheme 2 Top: Species found at end of Me ester reduction determined by 1H NMR using 1,4-dimethoxybenzene as internal standard-see ESI.† Below: Mechanistic proposal for ester transfer hydrogenation using 2 and EtOH. | ||
In ref. 5d, essentially no activity was described for a Mn/P,N,P catalyst in ester transfer hydrogenation. We believe catalysts 1–3 are significantly more active ester hydrogenation catalysts than the catalyst used in ref. 5d; indeed other researchers have compared P,N,N and P,N,P/Mn catalysts and found the former more reactive.12 In addition it is possible that the formation of H2 with a Mn/P,N,P catalyst13 could reduce the lifetime of the Mn-hydride needed to reduce the ester and hence the transfer hydrogenation efficiency. The presence of more N-donors might facilitate dehydrogenation without dihydrogen formation. One P-donor is still seemingly required for the hydride to be reactive enough to reduce an ester, as evidenced by the lack of reactivity of a catalyst derived from a simple bidentate N,N ligand, 5 (Table 1, entry 5). This catalyst gives high turnover numbers in ketone transfer hydrogenation,8 but we are not aware of ester pressure hydrogenations with any metal centre, using only pure N,N ligands. In summary, we have developed the first example of a manganese-catalysed reaction for the transfer hydrogenation of esters. The methodology operates using only 1 mol% of a sustainable metal catalyst to effect reduction.
We would like to thank the University of St Andrews, and the EPSRC Centre for Doctoral Training in Critical Resource Catalysis (CRITICAT) for financial support [PhD studentship to CO; Grant code: EP/L016419/1]. We thank Solvias for ongoing collaboration and the gift of chemicals.
Conflicts of interest
There are no conflicts to declare.Notes and references
-
(a) (Review): M. L. Clarke, Catal. Sci. Technol., 2012, 2, 2418–2423 RSC
; (b) (Review) S. Werkmeister, K. Junge and M. Beller, Org. Process Res. Dev., 2014, 18, 289–302 CrossRef CAS
- Fe-
(a) S. Chakraborty, H. Dai, P. Bhattacharya, N. T. Fairweather, M. S. Gibson, J. A. Krause and H. Guan, J. Am. Chem. Soc., 2014, 136, 7869–7872 CrossRef CAS PubMed
- M. B. Widegren and M. L. Clarke, Org. Lett., 2018, 20, 2654–2658 CrossRef CAS PubMed
-
(a) S. Gladiali and E. Alberico, Chem. Soc. Rev., 2006, 35, 226–236 RSC
-
(a) S.-H. Lee and G. I. Nikonov, ChemCatChem, 2015, 7, 107–113 CrossRef CAS
- D. R. Abernethy, A. J. DeStefano, T. L. Cecil, K. Zaidi, R. L. Williams and USP Metal Impurities Advisory Panel, Pharm. Res., 2010, 27, 750–755 CrossRef CAS PubMed
- M. B. Widegren and M. L. Clarke, Catal. Sci. Technol., 2019, 9, 6047–6058 RSC
- A. Bruneau-Voisine, D. Wang, V. Dorcet, T. Roisnel, C. Darcel and J.-B. Sortais, Org. Lett., 2017, 19, 3656–3659 CrossRef CAS PubMed
- M. A. Sánchez, G. C. Torres, V. A. Mazzieri and C. L. Pieck, J. Chem. Tech. Biotechnol., 2017, 92, 27–42 CrossRef
- Ru-
(a) T. vom Stein, M. Meuresch, D. Limper, M. Schmitz, M. Hölscher, J. Coetzee, D. J. Cole-Hamilton, J. Klankermayer and W. Leitner, J. Am. Chem. Soc., 2014, 136, 13217–13225 CrossRef CAS PubMed
- B. Neises and W. Steglich, Angew. Chem., Int. Ed. Engl., 1978, 17, 522–524 CrossRef
- Y. Wang, L. Zhu, Z. Shao, G. Li, Y. Lan and Q. Liu, J. Am. Chem. Soc., 2019, 141, 17337–17349 CrossRef CAS PubMed
- D. H. Nguyen, X. Trivelli, F. Capet, J.-F. Paul, F. Dumeignil and R. M. Gauvin, ACS Catal., 2017, 7, 2022–2032 CrossRef CAS
Footnotes |
| † Electronic supplementary information (ESI) available: Full experimental procedures, analytical data, NMR spectra. See DOI: 10.1039/d0cc02598d |
| ‡ The research data underpinning this publication can be accessed at https://doi.org/10.17630/0a645625-3a7a-47ed-9f6d-63ef3b998d79. |
| § Throughout this work, we have used single enantiomer catalysts since we had large amounts of these available, but we note ref. 7 shows, as one would expect, similar results using either single enantiomer or racemic catalysts in pressure hydrogenation. |
| This journal is © The Royal Society of Chemistry 2020 |