
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThree colour solid-state luminescence from positional isomers of facilely modified thiophosphoranyl anthracenes†
Timo
Schillmöller
,
Paul Niklas
Ruth
,
Regine
Herbst-Irmer
 and
Dietmar
Stalke
*
and
Dietmar
Stalke
*
Institut für Anorganische Chemie, Georg-August-Universität Göttingen, Tammannstraße 4, 37077 Göttingen, Germany. E-mail: dstalke@chemie.uni-goettingen.de
First published on 25th May 2020
Abstract
Three positional isomers of thiophosphoranyl anthracene were synthesized and their photophysical properties were investigated. By varying the position of the substituents, blue, green and yellow solid-state fluorescence with differences in the emission wavelength of over 100 nm, assigned to the intra- and intermolecular effects, could be established.
Weak intermolecular interactions play a key role in understanding the photophysical properties of luminescent solid-state materials.1,2 The traditional view of the Aggregation-Caused-Quenching (ACQ), which ascribes the commonly observed fluorescence quenching of the highly solution-emissive fluorophores in the solid state to the strong intermolecular interactions, has been overhauled.3 In recent years many concepts and strategies have been developed for tuning and improving the solid-state emission of organic molecules, which has been classically achieved by using metal-based compounds.4 The most famous concept is known as Aggregation-Induced-Emission (AIE) and comprises compounds that are non or only barely luminescent in solution but emit upon aggregation.5,6 Typically, the AIE-luminogens consist of a fluorophore substituted with a group, able to actively rotate or vibrate in solution, causing efficient quenching. When these compounds aggregate the internal motion is restricted and an enhanced solid-state emission is obtained.7 Tetraphenylethylene8 (TPE) and hexaphenylsilole5,9 (HPS) are the commonly used groups in the AIE-materials and a plethora of AIE-type fluorophores have already been investigated. Further approaches try to overcome the ACQ by introducing bulky substituents to the fluorophore or by co-crystal formation yielding less interchromophoric interactions and enhanced solid-state emission that originates from the monomeric form of the fluorophore.1,10 More recently, efficient solid-state emitters have been reported, which reveal a dimeric motif with the strong π–π interactions of the polyaromatic fluorophores resulting in an intense excimer emission.11 This way large bathochromic shifts of the emission wavelengths could be obtained. For the anthracene fluorophore, which usually emits in the blue region and can be used for photon up-conversion via a triplet–triplet annihilation,12 efficient solid-state fluorescence in the green region with the emission wavelengths up to 525 nm was achieved.13–15 Generally, the anthracene scaffold is substituted with a suitable group in the reactive 9-position and the weak interactions of the aromatic moieties result in a dimeric motif in the solid-state, which is suitable for excimer emission.13–15 In this paper we present the positional isomers of thiophosphoranyl anthracene. By substituting the uncommon 1- and 2-positions of the anthracene, solid-state emission in three colours with differences in the emission wavelength of more than 100 nm was observed. Surprisingly, a yellow excimer emission from the 1-isomer was observed, which is quite a rare colour for the structurally simple anthracene derivatives. Therefore, modifications in the 1-position of anthracene could be a promising strategy for obtaining a longer wavelength emission in the solid-state.
Herein, we report the synthesis and investigation of the structure–property relationship of the two positional isomers of the previously investigated [9-(S)PPh2(C14H9)]16 (3). [2-(S)PPh2(C14H9)] (2) was prepared from 2-bromoanthracene similar to the reported procedure for the 9-substituted derivative by the lithiation and the subsequent addition of chlorodiphenylphosphane.17 Oxidation of P(III) was achieved with elemental sulfur in toluene. [1-(S)PPh2(C14H9)] (1) was synthesized from 1-chloroanthracene by the nucleophilic substitution with lithium diphenylphosphanide and the subsequent sulfur oxidation. Shifting the substituent from the 9-position to the outer anthracene ring leads to a small downfield shift of the 31P-NMR resonance from about 35 ppm (3) to 42–43 ppm (1, 2), which is more typical for the thiophosphoranyl substituted aromatic compounds.16d,18 Furthermore, the oxidation leads to an increased stability compared to the P(III) precursors, which can undergo a slow oxidation in air.
Single crystals of 1 and 2 for structure determination were obtained by recrystallization from toluene. Both compounds crystallise in a monoclinic crystal system in the space groups P21/c (1) and P21/n (2). The solid-state structure of 3 has been reported earlier15 and reveals a triclinic crystal system in the space group P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) with two molecules in the asymmetric unit. As anticipated, 1 and 2 reveal the nearly planar anthracene moieties, in contrast to the strongly butterfly-bent structure of 3, with a folding angle α of 11.67(13)° and 16.86(11)°, respectively (see Fig. 1, Fig. S1, S2 (ESI†) and Table 1). Locating the bulky substituent at the outer anthracene ring induces less steric strain and a different conformation of the substituent regarding the anthracene plane. While in 3 both the phenyl groups are located above the anthracene plane resulting in an S–P–C–C torsion angle ω of 80.84(10)° and 89.01(10)°, in 1 and 2 a decrease of ω (1: 64.06(14)°, 2: 4.3(3)°) together with a changed substituent orientation is observed. Due to the more flexible orientation in 1 and 2 no bending of the anthracene is observed. Furthermore, the CAnth–P bond is slightly shortened and the CPh–P–CPh angle γ is increased for 1 and 2 indicating a less strained structure.
with two molecules in the asymmetric unit. As anticipated, 1 and 2 reveal the nearly planar anthracene moieties, in contrast to the strongly butterfly-bent structure of 3, with a folding angle α of 11.67(13)° and 16.86(11)°, respectively (see Fig. 1, Fig. S1, S2 (ESI†) and Table 1). Locating the bulky substituent at the outer anthracene ring induces less steric strain and a different conformation of the substituent regarding the anthracene plane. While in 3 both the phenyl groups are located above the anthracene plane resulting in an S–P–C–C torsion angle ω of 80.84(10)° and 89.01(10)°, in 1 and 2 a decrease of ω (1: 64.06(14)°, 2: 4.3(3)°) together with a changed substituent orientation is observed. Due to the more flexible orientation in 1 and 2 no bending of the anthracene is observed. Furthermore, the CAnth–P bond is slightly shortened and the CPh–P–CPh angle γ is increased for 1 and 2 indicating a less strained structure.
 | ||
| Fig. 1 Crystal structures of thiophosphoranyl anthracenes 1 (left), 2 (middle) and 3 (right) from the front view (top) and side view (bottom). For 3 the quantification of the anthracene bending is shown by the folding angle α. | ||
| 1 | 2 | 3 | |
|---|---|---|---|
| CAnth–P/Å | 1.8135(17) | 1.812(2) | 1.8326(13) |
| 1.8327(13) | |||
| γ/° | 104.62(8) | 106.58(10) | 101.65(6) |
| 100.95(6) | |||
| ω/° | 64.06(14) | 4.3(3) | 80.84(10) |
| 89.91(10) | |||
| α/° | 1.4(2) | 3.50(19) | 11.67(13) |
| 16.86(11) | |||
| Overlap/% | 42.3 | 0 | 21.1 |
| dπ–π/Å | 3.325 | — | 3.242 |
| dx/Å | 1.070 | — | 1.344 |
| dy/Å | 1.057 | — | 3.227 |
| λ em/nm | 545 | 426/441/469/502 | 484 |
| τ/ns | 29.0 | 3.2/7.8 | 1.4/3.2 |
| Φ F/% | 5.9 | 15.7 | 4.0 |
The UV-Vis absorption spectra of 1–3 reveal a structured absorption band from 340 to 410 nm, which is assigned to the S0 → S1 transition mainly located on the anthracene core and polarized along the short anthracene axis (see Fig. 2). Therefore, 1 and 3 show a slightly stronger red shift compared to 2 as the substituent is located on the short molecular axes. For 3 the vibronic structure is less pronounced due to the strong distortion of the anthracene plane. The emission spectra of 1 and 2 in diluted solution are nearly identical and also resemble the structure of the parent anthracene with a vibronic emission from 390 to 460 nm. The emission maximum of 3 is further red-shifted to 462 nm and the vibronic structure is completely lost, which is assigned to the bent structure of the anthracene.16d The emission in solution is generally weak due to a photoinduced electron transfer from the sulfur lone pair to the anthracene, which results in efficient quenching (see Table S4, ESI†). The emission intensity enhances when the aggregation is forced by the addition of different water fractions to the THF solution (see Fig. S13–S15, ESI†). Furthermore, a slight red-shift and a broadening of the spectra are observed at higher water fractions. When going to the solid-state, the emission properties change dramatically (see Fig. 3 and Table 1). For 2, the changes are only small and as in solution a blue emission can be observed in the range from 420 to 510 nm. The structured emission is still present even less pronounced compared to the diluted solution. For 3 also, a further red-shift of 20 nm compared to the solution emission is found, resulting in a blue-green emission in the solid-state. The shape of the spectrum is comparable with the structureless emission band in solution. For 1, the emission changes drastically upon aggregation. In the solid-state, a yellow emission with a wavelength of 545 nm is obtained. Compared to the solution spectra, the emission wavelength reveals a bathochromic shift of over 100 nm. Furthermore, the vibronic structure is completely lost and a largely broadened emission spectrum that ranges over 200 nm is obtained in the solid-state. Solid-state luminescence in the yellow region is quite rare for small anthracene derivatives without any conjugated substituent. As already mentioned, the hitherto reported anthracene excimers showed emission in the green region with a maximum wavelength of about 525 nm. The three positional isomers of the thiophosphoranyl anthracene reveal three colour emission in the solid-state with a difference in the emission wavelength of over 100 nm. The electronic structure of all three isomers should be comparable as the absorption and emission spectra in solution differ only slightly. Therefore, a closer look into the solid-state structures of 1–3 is needed to explain the large deviations in the emission spectra in the aggregated state.
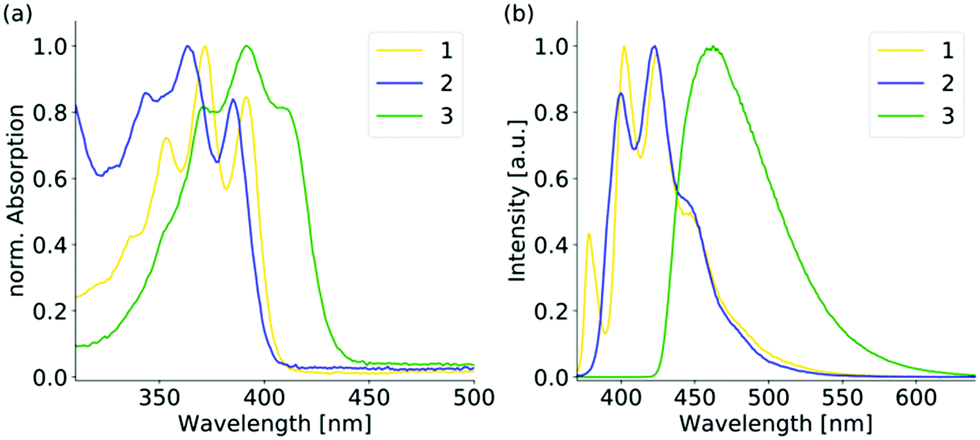 | ||
| Fig. 2 (a) Normalized UV-vis and (b) normalized emission spectra of the positional isomers 1–3 in the diluted THF solution (10−5 M, λex = 350 nm). | ||
 | ||
| Fig. 3 (a) Normalized solid-state emission spectra of the positional isomers and (b) photographs of the bulk samples of 1–3 under daylight (top) and under UV-light (bottom). | ||
Isomer 2 reveals the smallest change in the emission spectra upon aggregation. Both the solution and solid-state fluorescence resemble the emission of the unsubstituted anthracene. The solid-state structure is also closely related to pure anthracene.
No face-to-face interactions between the anthracene scaffolds can be observed and the closest interactions of the two anthracene cores are found in an end-to-face manner with C–H⋯π distances of around 2.8 Å (see Fig. 4). Further interactions are present between the phenyl groups of the backbone and the neighbouring anthracenes with similar C–H⋯π distances (see Fig. S4, ESI†). Through these general weak interactions towards the chromophore, the emission spectra are barely affected upon aggregation. For isomer 3 with the substituent located in the 9-position at the central ring, changes in the emission spectra are already seen in solution. The loss of the vibronic structure and the larger bathochromic shift has been ascribed to the strong folding and deformation of the anthracene plane. In the solid-state a further bathochromic shift was found, which has been attributed to the moderate face-to-face interactions between the two anthracenes resulting in a green emission. The mean π–π distance of these anthracene dimers is about 3.242 Å and the overlap was determined to be only 21.1%. As the broadened signal is already observed in solution and the lifetime of the fluorescence is only a few nanoseconds, an excimer formation is not assumed, and the emission is mainly attributed to the monomeric form of 3. The unusual large bathochromic shift of 1 resulting in a yellow emission is assigned to the strong intermolecular interactions. The dimeric motif of 3 is also dominant in the solid-state structure of 1 (see Fig. 5). The overlap is enlarged to about 42.3% as the offset dy along the long-molecular anthracene axes is decreased by over 2 Å to 1.057 Å compared to 3. The π–π distance with 3.325 Å is only slightly elongated. The shift of the substituent from the central to the outer anthracene ring leads to a new orientation of the substituent and therefore a less shielded anthracene site, which allows a larger overlap with less offset of the two anthracenes and consequently stronger π–π interactions. Therefore, the solid-state structure of 1 seems suitable for the formation of an excimer. In comparison to 3 the changes in 1 upon aggregation are much more pronounced. The emission spectrum is extremely broadened, and the bathochromic shift reaches nearly 100 nm. Together with the increased lifetime to almost 30 ns, an excimer formation as the origin of the yellow fluorescence of 1 can be confirmed. Besides the dimeric motif, further end-to-face interactions between anthracenes are observed, which lead to a ladder-type stacking with overall strong intermolecular interactions (see Fig. 5 and Fig. S6, ESI†). The quantum yields increase in the solid-state as the quenching by the sulfur lone pair is probably less effective due to the more rigid structure. Future research will focus on the introduction of different substituents to increase the emission efficiency and potentially leading to stronger overlaps and even more shifted emission wavelengths.
 | ||
| Fig. 4 Crystal packing of [2-(S)PPh2(C14H9)] (2) showing a herringbone-type packing of the anthracene moieties (bold). View along the crystallographic a-axis (a) and along crystallographic c-axis (b). Disorder is omitted for clarity. | ||
 | ||
| Fig. 5 (a) Crystal packing of [1-(S)PPh2(C14H9)] (1) revealing the strong face-to-face and end-to-face interactions of the anthracenes (bold). (b) Detailed view on the observed dimeric motif with a decent overlap of the anthracene planes. | ||
In conclusion, we have presented three positional isomers of a thiophosphoranyl anthracene that reveal solid-state fluorescence in three different colours with differences in emission wavelengths of over 100 nm. The analyses of the solid-state structures and the photophysical properties could ascribe the unusual yellow emission to the formation of an excimer in the solid-state. Therefore, substitution in the 1-position of the common anthracene fluorophore with suitable substituents could be a promising strategy for obtaining long-wavelength emission in the solid-state by using structurally easy to modify compounds.‡
This work was funded by the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) – 389479699/GRK2455.
Conflicts of interest
There are no conflicts to declare.Notes and references
- T. Hinoue, Y. Shigenoi, M. Sugino, Y. Mizobe, I. Hisaki, M. Miyata and N. Tohnai, Chem. – Eur. J., 2012, 18, 4634–4643 CrossRef CAS PubMed.
- (a) P. Srujana, P. Sudhakar and T. P. Radhakrishnan, J. Mater. Chem. C, 2018, 6, 9314–9329 RSC; (b) P.-Y. Gu, G. Liu, J. Zhao, N. Aratani, X. Ye, Y. Liu, H. Yamada, L. Nie, H. Zhang, J. Zhu, D.-S. Li and Q. Zhang, J. Mater. Chem. C, 2017, 5, 8869–8874 RSC; (c) Q. Li and Z. Li, Adv. Sci., 2017, 4, 1600484 CrossRef PubMed; (d) C. Wang and Z. Li, Mater. Chem. Front., 2017, 1, 2174–2194 RSC; (e) S. Varghese and S. Das, J. Phys. Chem. Lett., 2011, 2, 863–873 CrossRef CAS PubMed.
- (a) J. B. Birks, Photophysics of aromatic molecules, Wiley-Interscience, London, 1970 Search PubMed; (b) K. Shirai, M. Matsuoka and K. Fukunishi, Dyes Pigm., 1999, 42, 95–101 CrossRef CAS; (c) J. Gierschner, L. Lüer, B. Milián-Medina, D. Oelkrug and H.-J. Egelhaaf, J. Phys. Chem. Lett., 2013, 4, 2686–2697 CrossRef CAS; (d) L. Zong, Y. Xie, C. Wang, J.-R. Li, Q. Li and Z. Li, Chem. Commun., 2016, 52, 11496–11499 RSC.
- (a) J. N. Zhang, H. Kang, N. Li, S. M. Zhou, H. M. Sun, S. W. Yin, N. Zhao and B. Z. Tang, Chem. Sci., 2017, 8, 577–582 RSC; (b) M. K. Bera, P. Pal and S. Malik, J. Mater. Chem. C, 2020, 8, 788–802 RSC; (c) S. Varughese, J. Mater. Chem. C, 2014, 2, 3499–3516 RSC; (d) S. Xue, X. Qiu, Q. Sun and W. Yang, J. Mater. Chem. C, 2016, 4, 1568–1578 RSC; (e) O. S. Wenger, Chem. Rev., 2013, 113, 3686–3733 CrossRef CAS PubMed; (f) H. Terraschke and C. Wickleder, Chem. Rev., 2015, 20, 11352–11378 CrossRef PubMed.
- J. Luo, Z. Xie, J. W. Y. Lam, L. Cheng, H. Chen, C. Qiu, H. S. Kwok, X. Zhan, Y. Liu, D. Zhu and B. Z. Tang, Chem. Commun., 2001, 1740–1741 RSC.
- (a) Y. Hong, J. W. Y. Lam and B. Z. Tang, Chem. Commun., 2009, 4332–4353 RSC; (b) Y. Hong, J. W. Y. Lam and B. Z. Tang, Chem. Soc. Rev., 2011, 40, 5361–5388 RSC; (c) J. Mei, N. L. C. Leung, R. T. K. Kwok, J. W. Y. Lam and B. Z. Tang, Chem. Rev., 2015, 115, 11718–11940 CrossRef CAS PubMed.
- (a) Y. Tu, J. Liu, H. Zhang, Q. Peng, J. W. Y. Lam and B. Z. Tang, Angew. Chem., 2019, 131, 15053–15056 CrossRef; (b) Y. Xu, F. Wang, X. Chen, Y. Liu, Z. Zhou and B. Teng, J. Phys. Chem. C, 2019, 123, 29379–29385 CrossRef CAS; (c) H. Zhang, J. Liu, L. Du, C. Ma, N. L. C. Leung, Y. Niu, A. Qin, J. Sun, Q. Peng, H. H. Y. Sung, I. D. Williams, R. T. K. Kwok, J. W. Y. Lam, K. S. Wong, D. L. Phillips and B. Z. Tang, Mater. Chem. Front., 2019, 3, 1143–1150 RSC; (d) D. Presti, L. Wilbraham, C. Targa, F. Labat, A. Pedone, M. C. Menziani, I. Ciofini and C. Adamo, J. Phys. Chem. C, 2017, 121, 5747–5752 CrossRef CAS; (e) J. Sturala, M. K. Etherington, A. N. Bismillah, H. F. Higginbotham, W. Trewby, J. A. Aguilar, E. H. C. Bromley, A.-J. Avestro, A. P. Monkman and P. R. McGonigal, J. Am. Chem. Soc., 2017, 139, 17882–17889 CrossRef CAS PubMed.
- (a) H. Tong, Y. Hong, Y. Dong, M. Häussler, J. W. Y. Lam, Z. Li, Z. Guo, Z. Guo and B. Z. Tang, Chem. Commun., 2006, 3705–3707 RSC; (b) D. D. La, S. V. Bhosale, L. A. Jones and S. V. Bhosale, ACS Appl. Mater. Interfaces, 2018, 10, 12189–12216 CrossRef CAS PubMed; (c) Z. Yang, Z. Chi, Z. Mao, Y. Zhang, S. Liu, J. Zhao, M. P. Aldred and Z. Chi, Mater. Chem. Front., 2018, 2, 861–890 RSC.
- (a) G. Yu, S. Yin, Y. Liu, J. Chen, X. Xu, X. Sun, D. Ma, X. Zhan, Q. Peng, Z. Shuai, B. Tang, D. Zhu, W. Fang and Y. Luo, J. Am. Chem. Soc., 2005, 127, 6335–6346 CrossRef CAS PubMed; (b) M. Wang, G. Zhang, D. Zhang, D. Zhu and B. Z. Tang, J. Mater. Chem., 2010, 20, 1858–1867 RSC; (c) Z. Zhao, B. He and B. Z. Tang, Chem. Sci., 2015, 6, 5347–5365 RSC.
- (a) R. Bhowal, S. Biswas, A. Thumbarathil, A. L. Koner and D. Chopra, J. Phys. Chem. C, 2019, 123, 9311–9322 CrossRef CAS; (b) Y. Huang, J. Xing, Q. Gong, L.-C. Chen, G. Liu, C. Yao, Z. Wang, H.-L. Zhang, Z. Chen and Q. Zhang, Nat. Commun., 2019, 10, 169 CrossRef PubMed; (c) R. Usman, A. Khan, M. Wang, Y. Luo, W. Sun, H. Sun, C. Du and N. He, Cryst. Growth Des., 2018, 18, 6001–6008 CrossRef CAS.
- (a) W. Jiang, Y. Shen, Y. Ge, C. Zhou, Y. Wen, H. Liu, H. Liu, S. Zhang, P. Lu and B. Yang, J. Mater. Chem. C, 2020, 8, 3367–3373 RSC; (b) Y. Shen, H. Liu, J. Cao, S. Zhang, W. Li and B. Yang, Phys. Chem. Chem. Phys., 2019, 21, 14511–14515 RSC; (c) Y. Shen, Z. Zhang, H. Liu, Y. Yan, S. Zhang, B. Yang and Y. Ma, J. Phys. Chem. C, 2019, 123, 13047–13056 CAS.
- (a) C. Kerzig and O. S. Wenger, Chem. Sci., 2018, 9, 6670–6678 RSC; (b) M. Xu, C. Han, Y. Yang, Z. Shen, W. Feng and F. Li, J. Mater. Chem. C, 2016, 4, 9986–9992 RSC; (c) R. Tao, J. Zhao, F. Zhong, C. Zhang, W. Yang and K. Xu, Chem. Commun., 2015, 51, 12403–12406 RSC.
- H. Liu, D. Cong, B. Li, L. Ye, Y. Ge, X. Tang, Y. Shen, Y. Wen, J. Wang, C. Zhou and B. Yang, Cryst. Growth Des., 2017, 17, 2945–2949 CrossRef CAS.
- H. Liu, L. Yao, B. Li, X. Chen, Y. Gao, S. Zhang, W. Li, P. Lu, B. Yang and Y. Ma, Chem. Commun., 2016, 52, 7356–7359 RSC.
- Y. Shen, H. Liu, S. Zhang, Y. Gao, B. Li, Y. Yan, Y. Hu, L. Zhao and B. Yang, J. Mater. Chem. C, 2017, 5, 10061–10067 RSC.
- (a) C. Köhler, J. Lübben, L. Krause, C. Hoffmann, R. Herbst-Irmer and D. Stalke, Acta Crystallogr., 2019, 424–441 Search PubMed; (b) L. Krause, B. Niepötter, C. J. Schürmann, D. Stalke and R. Herbst-Irmer, IUCrJ, 2017, 4, 420–430 CrossRef CAS PubMed; (c) B. Niepötter, R. Herbst-Irmer and D. Stalke, J. Appl. Crystallogr., 2015, 48, 1485–1497 CrossRef; (d) A. T. Breshears, A. C. Behrle, C. L. Barnes, C. H. Laber, G. A. Baker and J. R. Walensky, Polyhedron, 2015, 100, 333–343 CrossRef CAS; (e) R. Herbst-Irmer, J. Henn, J. J. Holstein, C. B. Hübschle, B. Dittrich, D. Stern, D. Kratzert and D. Stalke, J. Phys. Chem. A, 2013, 117, 633–641 CrossRef CAS PubMed.
- (a) Z. Fei, N. Kocher, C. J. Mohrschladt, H. Ihmels and D. Stalke, Angew. Chem., Int. Ed., 2003, 42, 783–787 CrossRef CAS PubMed; (b) G. Schwab, D. Stern, D. Leusser and D. Stalke, Z. Naturforsch. B, 2007, 62, 711–716 CAS; (c) D. Stern, N. Finkelmeier, K. Meindl, J. Henn and D. Stalke, Angew. Chem., Int. Ed., 2010, 49, 6869–6872 CrossRef CAS PubMed; (d) N. Finkelmeier, A. Visscher, S. Wandtke, R. Herbst-Irmer and D. Stalke, Chem. Commun., 2016, 52, 5440–5442 RSC.
- (a) G. Baccolini, C. Boga and M. Mazzacurati, J. Org. Chem., 2005, 70, 4774–4777 CrossRef CAS PubMed; (b) M. Hayashi, T. Matsuura, I. Tanaka, H. Ohta and Y. Watanabe, Org. Lett., 2013, 15, 628–631 CrossRef CAS PubMed.
- T. Schulz, K. Meindl, D. Leusser, D. Stern, J. Graf, C. Michaelsen, M. Ruf, G. M. Sheldrick and D. Stalke, J. Appl. Crystallogr., 2009, 42, 885–891 CrossRef CAS.
- L. Krause, R. Herbst-Irmer, G. M. Sheldrick and D. Stalke, J. Appl. Crystallogr., 2015, 48, 3–10 CrossRef CAS PubMed.
- L. Krause, R. Herbst-Irmer and D. Stalke, J. Appl. Crystallogr., 2015, 48, 1907–1913 CrossRef CAS.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Adv., 2015, 71, 3–8 CrossRef PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 3–8 Search PubMed.
- C. B. Hübschle and B. Dittrich, J. Appl. Crystallogr., 2011, 44, 238–240 CrossRef PubMed.
- D. Kratzert, J. J. Holstein and I. Krossing, J. Appl. Crystallogr., 2015, 48, 933–938 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. 1993261 and 1993262. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0cc02585b |
| ‡ Crystallographic details: crystallographic data were collected from a Mo-IμS microfocus source.19 All data were integrated with SAINT. A multiscan absorption correction (SADABS)20 and a 3λ correction21 were applied. The structures were solved by using direct methods (SHELXT)22 and refined on F2 using the full-matrix least-squares methods of SHELXL23 within the ShelXle GUI.24 Disordered groups were modelled using DSR.25 |
| This journal is © The Royal Society of Chemistry 2020 |