
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRegioselective biocatalytic self-sufficient Tishchenko-type reaction via formal intramolecular hydride transfer†
Erika
Tassano
 a,
Kemal
Merusic
a,
Isa
Buljubasic
a,
Olivia
Laggner
a,
Tamara
Reiter
a,
Andreas
Vogel
b and
Mélanie
Hall
*a
a,
Kemal
Merusic
a,
Isa
Buljubasic
a,
Olivia
Laggner
a,
Tamara
Reiter
a,
Andreas
Vogel
b and
Mélanie
Hall
*a
aDepartment of Chemistry, University of Graz, Heinrichstrasse 28, 8010 Graz, Austria. E-mail: melanie.hall@uni-graz.at
bc-LEcta GmbH, Perlickstrasse 5, 04103 Leipzig, Germany
First published on 28th April 2020
Abstract
A self-sufficient nicotinamide-dependent intramolecular bio-Tishchenko-type reaction was developed. The reaction is catalyzed by alcohol dehydrogenases and proceeds through formal intramolecular hydride transfer on dialdehydes to deliver lactones. Regioselectivity on [1,1′-biphenyl]-2,2′-dicarbaldehyde substrates could be controlled via the electronic properties of the substituents. Preparative scale synthesis provided access to substituted dibenzo[c,e]oxepin-5(7H)-ones.
The Claisen–Tishchenko reaction is a disproportionation reaction that leads to the formation of esters through dimerization of aldehydes.1 Due to the variety of protocols available, it is a synthetically relevant transformation2 and one of the major approaches for the bulk production of ethyl acetate.3 The Lewis acid promoted Tishchenko reaction involves a reduction–oxidation sequence, typically in the presence of aluminium alkoxides.2 The intramolecular reaction transforms dialdehydes to corresponding lactones, and several catalysts – mostly transition metal and rare earth compounds – have been explored with varying efficiency.4–6 1,4- and 1,5-dialdehydes are preferred since formation of γ- and δ-lactones, respectively, is thermodynamically favored. The reaction proceeds through hydrogen or hydride borrowing,7,8 whereby one aldehyde is first reduced to the alcohol, which combines with the second aldehyde group to yield a hemiacetal intermediate that gets oxidized. This redox-neutral reaction can be applied to ketoaldehydes.5,8,9 To the best of our knowledge, biocatalytic equivalents have not been disclosed. The case of photoheterotrophic cultures of the algae Euglena gracilis Z, which were reported to promote the conversion of o-phthalaldehyde to phthalide,10 raised our interest, as it indicates that a biocatalytic approach to the intramolecular Tishchenko reaction may be possible. No clear mechanistic pathway was identified, but elusive formation of 2-(hydroxymethyl)-benzoic acid as intermediate was suggested.
Recently, we established a biocatalytic variant of the (intermolecular) Cannizzaro reaction, which could be further developed into a parallel interconnected dynamic asymmetric disproportionation of α-substituted aldehydes. The system relies on the concurrent oxidative and reductive activity of nicotinamide-dependent alcohol dehydrogenases (ADHs) to perform the dismutation of aldehydes with high enantioselectivity in a redox-neutral fashion.11,12 The latter feature renders the system appealing from a sustainability point of view, since only catalytic amounts of cofactor are necessary, in addition to the biocatalyst. Since two products are generated, an additional purification step is necessary, or alternatively, convergent processes toward a single product must be designed. In this regard, the intramolecular Tishchenko reaction is highly attractive since only one product may be formed.
Pioneering work by Jones and coworkers showed that the enzymatic double oxidation of 1,4- and 1,5-diols catalyzed by NAD-dependent horse liver alcohol dehydrogenase (HLADH) proceeds through formation of the hydroxyaldehyde that spontaneously undergoes cyclization to the corresponding lactol, which gets further oxidized to the lactone product; overall two equivalents of NAD+ are necessary.13,14 The HLADH-catalyzed transformation of diols to lactones was subsequently employed in studies focusing on nicotinamide cofactor regeneration strategies.15 This biotransformation recently witnessed a ‘renaissance’16 and has been applied to various synthetic and cofactor recycling protocols.17 Biocatalytic systems based on laccase-catalyzed oxidation of diols to lactones also exist.18
Aiming at high hydride-economy in biosynthetic protocols, we evaluated the feasibility of a nicotinamide-dependent self-sufficient biocatalytic intramolecular Tishchenko-type reaction catalyzed by a single ADH in presence of catalytic amounts of cofactor, which would not rely on the presence of a co-substrate nor generate any by-product (Scheme 1). Such minimalistic in vitro cofactor-dependent biocatalytic systems yielding a single product are rare,16 and so far limited to racemization of enantiomerically pure molecules19 and redox isomerization reaction.20
 | ||
| Scheme 1 Biocatalytic intramolecular Tishchenko-like reaction catalyzed by alcohol dehydrogenase (ADH). | ||
The catalytic activity of several ADHs with dialdehydes toward formation of lactone products was first investigated on o-phthalaldehyde (1a) as model substrate, employing a collection of NADH-dependent ADHs (Scheme 2).21 All reactions were run in presence of acetonitrile as co-solvent in a concentration typically well tolerated by ADHs (5 vol%).16,17,26b Catalytic amounts of cofactor (0.05 eq.) were employed on 10 mM substrate and the reaction was run at pH 7 and 30 °C. Phthalide (1b) was obtained as sole product with several enzymes in up to 74% conversion, indicating internal recycling of the cofactor (up to ∼15 turnovers, see ESI,† Table S1) and effective formal intramolecular 1,4-hydride shift. Accumulation of the lactol intermediate could be detected only in a few cases (see ESI,† Fig. S1). This confirms that the reaction proceeds through a reduction–oxidation sequence, with formation of the hydroxyaldehyde first – leading to the lactol13 – followed by its oxidation to the lactone (Scheme 1). No product was formed in absence of enzyme. The enzymatic reaction proceeded fast on 1a (max. conversion reached after ∼120 min with ADH-172, see ESI,† Fig. S2) and additional experiments conducted with ADH-132 seem to indicate that conversion was not limited by the activity of the enzyme but rather by the substrate and/or cofactor availability (see ESI,† Table S2).
 | ||
| Scheme 2 Conversion of 1a–10a into two possible regioisomers 1b–10b and 1c–10c in the intramolecular Tishchenko-like reaction catalyzed by alcohol dehydrogenase (ADH). The cofactor NAD(P)H is added in catalytic amounts. | ||
The scope of the reaction was enlarged by testing structurally related 2a and 3a (see ESI,† Table S3). Since these dialdehydes are unsymmetrical, the reaction can produce two regioisomers 2/3b and 2/3c (Scheme 2), depending on the regioselectivity of the biocatalyst. In general, 2-(3-oxopropyl)benzaldehyde (3a) was better accepted by most ADHs, and conversion reached up to 70% with ADH-110 (entry 3, Table S3, ESI†). In contrast, conversion of 2-(2-oxoethyl)benzaldehyde (2a) was limited to max. 27% with ADH-171 (entry 11, Table S3, ESI†). The regiopreference was strongly enzyme-dependent. With 3a, in all cases except with ADH-170, 4,5-dihydrobenzo[c]oxepin-1(3H)-one (3b) was the major product. Noteworthy, ADH-114 displayed exquisite regioselectivity (3b/3c in >99![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio), along with very good conversion level, which was at the highest on 20 mM 3a (added in two aliquots over 3 h, 67% conversion, entry 6, Table S3, ESI†). Complementary biocatalytic approach to such lactones using Baeyer–Villiger monooxygenase could not produce this regioisomer.22 On 2a, the regioselectivity pattern was mixed, with enzymes displaying moderate conversion level and preference for isochroman-3-one (2c), or low conversion level along with preference for isochroman-1-one (2b). With ADH-110, 111 and 132, which did not produce any lactones 2b–2c from 2a, traces of lactol were detected, indicating that the oxidation step in these cases was not taking place. Since the reduced cofactor is used in catalytic amounts, the overall reaction relies on the regeneration of NADH through lactol oxidation. In its absence, the system reaches a dead-end and the substrate does not get converted beyond the availability of the cofactor, here 0.5 mM. Other competing phenomena with 2a, likely initiated by the hydration of one aldehyde group,11 were observed but not studied further (see ESI,† Fig. S3 and Scheme S1). A time study performed on 3a with ADH-114 highlighted that the reaction was much slower compared to the biotransformation of 1a (see ESI,† Fig. S4). This reaction was performed on 200 mg scale. Under the reaction conditions reported in Table S3 (ESI†) (entry 6), 105 mg of 3b could be obtained after purification by flash chromatography (53% isolated yield, see ESI,† Fig. S5).
:1 ratio), along with very good conversion level, which was at the highest on 20 mM 3a (added in two aliquots over 3 h, 67% conversion, entry 6, Table S3, ESI†). Complementary biocatalytic approach to such lactones using Baeyer–Villiger monooxygenase could not produce this regioisomer.22 On 2a, the regioselectivity pattern was mixed, with enzymes displaying moderate conversion level and preference for isochroman-3-one (2c), or low conversion level along with preference for isochroman-1-one (2b). With ADH-110, 111 and 132, which did not produce any lactones 2b–2c from 2a, traces of lactol were detected, indicating that the oxidation step in these cases was not taking place. Since the reduced cofactor is used in catalytic amounts, the overall reaction relies on the regeneration of NADH through lactol oxidation. In its absence, the system reaches a dead-end and the substrate does not get converted beyond the availability of the cofactor, here 0.5 mM. Other competing phenomena with 2a, likely initiated by the hydration of one aldehyde group,11 were observed but not studied further (see ESI,† Fig. S3 and Scheme S1). A time study performed on 3a with ADH-114 highlighted that the reaction was much slower compared to the biotransformation of 1a (see ESI,† Fig. S4). This reaction was performed on 200 mg scale. Under the reaction conditions reported in Table S3 (ESI†) (entry 6), 105 mg of 3b could be obtained after purification by flash chromatography (53% isolated yield, see ESI,† Fig. S5).
In addition to commercial ADHs, we turned our attention to a variant of NADPH-dependent ADH from Lactobacillus kefir (Lk-ADHmut), which was designed by Codexis to accept bulky–bulky substrates.23,24 The enzyme was cloned and over-expressed in E. coli and used in purified form (see ESI†). The enzyme showed very good activity on 2a–3a (up to 81% activity on 20 mM of 2a, entry 17, Table S3, ESI†), with pronounced regiopreference for 2b, but mixed selectivity on 3a, confirming the general trend of the tested ADHs not able to perform best on both substrates simultaneously, a surprising feature given the high structural similarity between 2a and 3a.
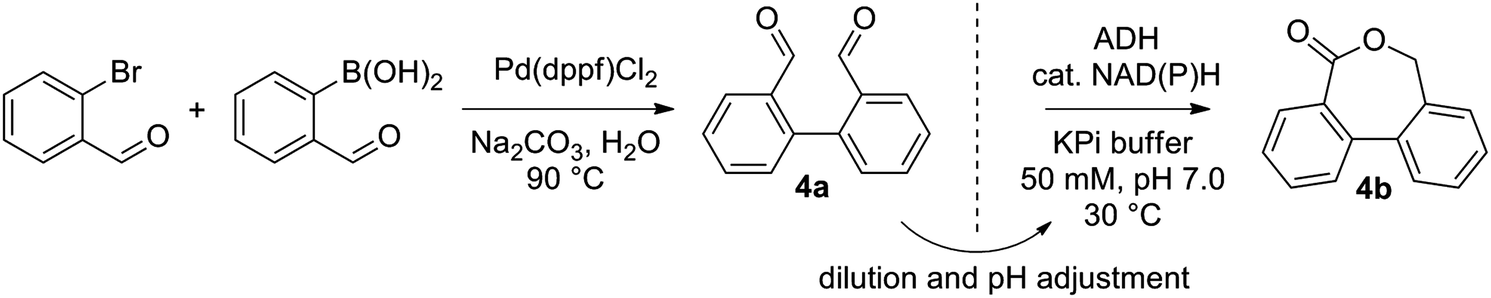
Next, a range of variously substituted [1,1′-biphenyl]-2,2′-dicarbaldehyde substrates 4a–10a obtained by Pd-catalyzed Suzuki–Miyaura coupling reaction25 was studied, aiming at the synthesis of dibenzo[c,e]oxepin-5(7H)-ones. Good to excellent conversion levels were obtained with non-substituted 4a, with Lk-ADHmut performing the best (82–90% conversion on 5–15 mM 4a, entry 1, Table 1, and see ESI,† Table S4). Inspired by previous reports demonstrating the compatibility of ADH with Pd-catalyst,26 we designed an aqueous chemoenzymatic one-pot two-step cascade for the synthesis of 4b on 50 mg scale (Scheme 3). The Pd-catalyzed cross-coupling reaction proceeded in water at 90 °C with 3 eq. of Na2CO3, and was followed – upon dilution to 15 mM 4a with buffer and 5 vol% acetonitrile and pH adjustment – by the ADH-catalyzed reaction using purified Lk-ADHmut, in presence of 0.03 eq. NADPH. After purification by flash chromatography, dibenzo[c,e]oxepin-5(7H)-one (4b) could be obtained in 48% isolated yield (entry 1, Table 1).
| Entry | Substrate | ADH | Conv. (%) | Ratio 4-10b/4-10c | Isolated yieldb (%) |
|---|---|---|---|---|---|
| a 10 mM of 4a–10a, 1 mg mL−1 ADH, KPi buffer (50 mM, pH 7.0, 2 mM MgCl2), 5 vol% CH3CN, 30 °C, 500 μL volume, 120 rpm. b 0.1–0.41 mmol scale. c One-pot two-step sequence from bromoaldehyde and 2-formylphenylboronic acid. d 3 mg mL−1 ADH, 1 mM NADH. e Lk-ADHmut employed due to better availability of the enzyme preparation. | |||||
| 1 | 4a | Lk-ADHmut | 84 | n.a. | 48c |
| 2 | 5a | 132d | 96 | >99:1 |
29 (5b) |
| 3 | 6a | Lk-ADHmut | 75 | 97:3 |
66 (6b) |
| 4 | 7a | Lk-ADHmut | 98 | 14:86 |
74 (7c) |
| 5 | 8a | 132 | 57 | 1:>99 |
28 (8c) |
| 6 | 9a | 118 | 52 | 1:>99 |
58e (9c) |
| 7 | 10a | 107/132 | >99 | n.a. | 56e |
 | ||
| Scheme 3 Aqueous sequential chemoenzymatic one-pot cascade of Suzuki–Miyaura coupling and ADH-catalyzed lactonization. | ||
During further study of the enzymatic reaction, the electronic properties of the substituents were found to have a major effect on the conversion levels of 5a–10a. Overall, the electron-withdrawing group (EWG, –F) strongly favored activity, as most enzymes showed good to excellent activity on 7a (up to 98% conversion, entry 4, Table 1), while the effect of the electron-donating group (EDG, –OMe) was opposite and most enzymes were poorly active or not active on 5a–6a (see ESI,† Table S5). With 5a–6a, ADH-132 showed promising conversion, which could be boosted on 5a by providing more enzyme and cofactor (entry 2, Table 1). Lk-ADHmut was found very active on 6a (entry 3, Table 1), however, conversion of 5a was very poor (5%, see ESI,† Table S5). More striking was the degree of regioselectivity observed with all enzymes, which preferentially delivered one regioisomer. Regardless of the biocatalyst, the EWG was always found on the ring bearing the aldehyde that got reduced first, leading to the formation of 8-fluorodibenzo[c,e]oxepin-5(7H)-one (7c), while the EDG was found on the ring bearing the aldehyde that remained inert in the first reductive step, leading to the formation of 4- and 2-methoxydibenzo[c,e]oxepin-5(7H)-ones (5b and 6b). Clearly, the EWG increased the electrophilicity of the aldehyde group and thereby favored attack by the hydride, while the EDG favored attack of the aldehyde group located on the opposite ring (Scheme S2, ESI†). The regiopreference was exquisite on 5a (no trace of 5c detected) and led to predominant formation of regioisomers 6b and 7c with 6a and 7a, respectively (up to 97:3 ratio for 6b/6c in 75% conversion and 5:95 ratio for 7b/7c in 15% conversion, respectively, see ESI,† Table S5). These effects were confirmed by adding a substituent on the second ring. Conversion of 10a was increased in most cases compared to conversion of 7a (up to full conversion, see ESI,† Tables S6, S7 and entry 7, Table 1), confirming that EWGs are facilitating the reaction. Addition of an EWG on poorly reactive 5a–6a turned beneficial in selected cases and led to significant increase in conversion level of 8a with some biocatalysts (up to 400% increase in conversion compared to 5a), while the effect was much less encouraging on 9a (see ESI,† Tables S6 and S7); with the latter, most dramatic change was seen in the regioselectivity, as all ADHs delivered only regioisomer 9c, which can hint as a ‘cumulative’ effect of the electronic properties on the regioselectivity (see ESI,† Table S6 and Scheme S2). Finally, these substrates were subjected to preparative synthesis on up to 100 mg scale and corresponding dibenzo[c,e]oxepin-5(7H)-ones 4b–6b and 7c–10c could be obtained after purification by flash chromatography in very good isolated yield (up to 74%, Table 1) and excellent purity (see ESI†).
In order to analyze the hydride economy and the overall catalytic performance of the system, use of isolated and purified ADH was necessary. To that end, and given the high conversion level achieved with 0.05 eq. NADPH and 1 mg mL−1 ADH (∼37 μM, 0.37 mol%), the transformation of 7a by Lk-ADHmut was closely looked at (Table 2). Cofactor and enzyme concentrations were varied and the resulting data indicate that (i) the catalyst was highly active and conversion up to 77% could be achieved using only 3.7 μM (0.037 mol%) of ADH, which corresponds to a total turnover number (TTN) of 2081 (entry 5, Table 2); (ii) the hydride economy was excellent: turnover number (TON) up to 880 was achieved with 0.001 eq. of NADPH, translating in 1.6 × 103 half-reactions per mole of cofactor (entry 2, Table 2). This cofactor loading is one of the lowest reported for NAD(P)-dependent self-sufficient biocatalytic systems yielding such high conversion levels.16 In biotransformations (one substrate → one product) using a combination of enzymes, max. 470 TON has been reported (0.002 eq. of NADPH)27 but typical TON values are <100.16 Compared to reported protocols relying on a single ADH (TON 10–30),11,19,20 the bio-Tischenko-like reaction stands out due to highly effective internal hydride shuffling, likely due to the thermodynamic advantage provided by the spontaneous cyclization of the hydroxyaldehyde.
Collectively, the data demonstrate that formal intramolecular 1,4-, 1,5- and 1,6-hydride shift on dialdehydes can be catalyzed by nicotinamide-dependent alcohol dehydrogenases. The resulting bio-Tishchenko-like reaction provides access to lactones in a redox-neutral manner through a simple protocol under mild conditions in aqueous environment (max. 5 vol% organic co-solvent). By investigating a panel of ADHs, regioselective formation of isochroman-1-one and 4,5-dihydrobenzo[c]oxepin-1(3H)-one could be achieved. With substituted [1,1′-biphenyl]-2,2′-dicarbaldehydes, regiocomplementarity could be induced by varying the electronic properties of the substituent. This strategy was successfully applied to the regioselective synthesis of 2-, 4- and 8-substituted dibenzo[c,e]oxepin-5(7H)-ones. Importantly, this self-sufficient biocatalytic system displays excellent hydride economy. Combination of the enzymatic step with Pd-catalyzed cross-coupling in one pot finally provides an elegant chemoenzymatic cascade applicable to the synthesis of lactones.
Funding by the Austrian Science Fund is gratefully acknowledged (P30519-N36). We thank Prof. K. Zangger and B. Werner for NMR measurements, P. M. Neu for HRMS analyses, A. Billiani and L. Schober for technical assistance, and J. Schrittwieser and M. Fuchs for fruitful scientific discussions.
Conflicts of interest
There are no conflicts to declare.References
- S. A. Morris and D. G. Gusev, Angew. Chem., Int. Ed., 2017, 56, 6228 CrossRef CAS PubMed.
- T. Seki, T. Nakajo and M. Onaka, Chem. Lett., 2006, 35, 824 CrossRef CAS.
- M. Nielsen, H. Junge, A. Kammer and M. Beller, Angew. Chem., Int. Ed., 2012, 51, 5711 CrossRef CAS PubMed.
- (a) S. H. Bergens, D. P. Fairlie and B. Bosnich, Organometallics, 1990, 9, 566 CrossRef CAS; (b) M. R. Burgstein, H. Berberich and P. W. Roesky, Chem. – Eur. J., 2001, 7, 3078 CrossRef CAS; (c) S. Onozawa, T. Sakakura, M. Tanaka and M. Shiro, Tetrahedron, 1996, 52, 4291 CrossRef CAS; (d) T. Ooi, T. Miura, K. Takaya and K. Maruoka, Tetrahedron Lett., 1999, 40, 7695 CrossRef CAS.
- J. Uenishi, S. Masuda and S. Wakabayashi, Tetrahedron Lett., 1991, 32, 5097 CrossRef CAS.
- T. Ooi, T. Miura, Y. Itagaki, I. Ichikawa and K. Maruoka, Synthesis, 2002, 279 CAS.
- (a) M. G. Edwards and J. M. J. Williams, Angew. Chem., Int. Ed., 2002, 41, 4740 CrossRef CAS PubMed; (b) A. Labonne, L. Zani, L. Hintermann and C. Bolm, J. Org. Chem., 2007, 72, 5704 CrossRef CAS PubMed; (c) M. C. Haibach and D. Seidel, Angew. Chem., Int. Ed., 2014, 53, 5010 CrossRef CAS PubMed.
- M. H. S. A. Hamid, P. A. Slatford and J. M. J. Williams, Adv. Synth. Catal., 2007, 349, 1555 CrossRef CAS.
- (a) J. L. Hsu and J. M. Fang, J. Org. Chem., 2001, 66, 8573 CrossRef CAS PubMed; (b) S. Omura, T. Fukuyama, Y. Murakami, H. Okamoto and I. Ryu, Chem. Commun., 2009, 6741 RSC; (c) Z. M. Shen, P. K. Dornan, H. A. Khan, T. K. Woo and V. M. Dong, J. Am. Chem. Soc., 2009, 131, 1077 CrossRef CAS PubMed; (d) Z. M. Shen, H. A. Khan and V. M. Dong, J. Am. Chem. Soc., 2008, 130, 2916 CrossRef CAS PubMed; (e) T. Suzuki, T. Yamada, K. Watanabe and T. Katoh, Bioorg. Med. Chem. Lett., 2005, 15, 2583 CrossRef CAS PubMed.
- Y. Noma, Y. Okajima, H. Takahashi and Y. Asakawa, Phytochemistry, 1991, 30, 2969 CrossRef CAS.
- E. Tassano, K. Faber and M. Hall, Adv. Synth. Catal., 2018, 360, 2742 CrossRef CAS PubMed.
- C. Wuensch, H. Lechner, S. M. Glueck, K. Zangger, M. Hall and K. Faber, ChemCatChem, 2013, 5, 1744 CrossRef CAS.
- A. J. Irwin and J. B. Jones, J. Am. Chem. Soc., 1977, 99, 1625 CrossRef CAS PubMed.
- (a) A. J. Irwin and J. B. Jones, J. Am. Chem. Soc., 1977, 99, 556 CrossRef CAS PubMed; (b) I. J. Jakovac, H. B. Goodbrand, K. P. Lok and J. B. Jones, J. Am. Chem. Soc., 1982, 104, 4659 CrossRef CAS; (c) J. B. Jones and C. J. Francis, Can. J. Chem./Rev. Can. Chim., 1984, 62, 2578 CrossRef.
- (a) G. Hilt, B. Lewall, G. Montero, J. H. P. Utley and E. Steckhan, Liebigs Ann./Recueil, 1997, 2289 CrossRef CAS; (b) I. Schroder, E. Steckhan and A. Liese, J. Electroanal. Chem., 2003, 541, 109 CrossRef CAS.
- E. Tassano and M. Hall, Chem. Soc. Rev., 2019, 48, 5596 RSC.
- (a) S. Gargiulo, D. J. Opperman, U. Hanefeld, I. W. C. E. Arends and F. Hollmann, Chem. Commun., 2012, 48, 6630 RSC; (b) S. Kara, D. Spickermann, J. H. Schrittwieser, C. Leggewie, W. J. H. van Berkel, I. W. C. E. Arends and F. Hollmann, Green Chem., 2013, 15, 330 RSC; (c) S. Kara, D. Spickermann, J. H. Schrittwieser, A. Weckbecker, C. Leggewie, I. W. C. E. Arends and F. Hollmann, ACS Catal., 2013, 3, 2436 CrossRef CAS; (d) R. Zuhse, C. Leggewie, F. Hollmann and S. Kara, Org. Process Res. Dev., 2015, 19, 369 CrossRef CAS.
- (a) A. Diaz-Rodriguez, L. Martinez-Montero, I. Lavandera, V. Gotor and V. Gotor-Fernandez, Adv. Synth. Catal., 2014, 356, 2321 CrossRef CAS; (b) A. Diaz-Rodriguez, I. Lavandera, S. Kanbak-Aksu, R. A. Sheldon, V. Gotor and V. Gotor-Fernandez, Adv. Synth. Catal., 2012, 354, 3405 CrossRef CAS.
- C. C. Gruber, B. M. Nestl, J. Gross, P. Hildebrandt, U. T. Bornscheuer, K. Faber and W. Kroutil, Chem. – Eur. J., 2007, 13, 8271 CrossRef CAS PubMed.
- Y. C. Liu, C. Merten and J. Deska, Angew. Chem., Int. Ed., 2018, 57, 12151 CrossRef CAS PubMed.
- R. Schmiedel, A. Vogel, S. Köpke, R. Czaja, C. Feller, H. Merkens, K. Rzeznicka, A. Petri, D. Schwarze, M. Struhalla and T. Greiner-Stöffele, WO 2015/162064 A1, 2015.
- A. Rioz-Martinez, G. de Gonzalo, D. E. T. Pazmino, M. W. Fraaije and V. Gotor, Eur. J. Org. Chem., 2009, 2526 CrossRef CAS.
- N. K. Modukuru, J. Sukumaran, S. J. Collier, A. S. Chan, A. Gohel, G. W. Huisman, R. Keledjian, K. Narayanaswamy, S. J. Novick, S. M. Palanivel, D. Smith, Z. Wei, B. Wong, W. L. Yeo and D. A. Entwistle, Org. Process Res. Dev., 2014, 18, 810 CrossRef CAS.
- A. Gohel, D. J. Smith, B. Wong, J. Sukumaran, W. L. Yeo, S. J. Collier and S. Novick, US Pat., US20140199735, 2014 Search PubMed.
- N. Miyaura and A. Suzuki, Chem. Rev., 1995, 95, 2457 CrossRef CAS.
- (a) E. Burda, W. Hummel and H. Groger, Angew. Chem., Int. Ed., 2008, 47, 9551 CrossRef CAS PubMed; (b) D. Gonzalez-Martinez, V. Gotor and V. Gotor-Fernandez, ChemCatChem, 2019, 11, 5800 CrossRef CAS.
- T. Knaus, F. G. Mutti, L. D. Humphreys, N. J. Turner and N. S. Scrutton, Org. Biomol. Chem., 2015, 13, 223 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0cc02509g |
| This journal is © The Royal Society of Chemistry 2020 |