
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Amphiphilic dendrimers control protein binding and corona formation on liposome nanocarriers†
Jessica
Wagner‡
ab,
Marcel
Dillenburger‡
a,
Johanna
Simon
ac,
Jennifer
Oberländer
ac,
Katharina
Landfester
 a,
Volker
Mailänder
ac,
David Y. W.
Ng
a,
Klaus
Müllen
*a and
Tanja
Weil
*a
a,
Volker
Mailänder
ac,
David Y. W.
Ng
a,
Klaus
Müllen
*a and
Tanja
Weil
*a
aMax Planck Institute for Polymer Research, Ackermannweg 10, 55128 Mainz, Germany. E-mail: muellen@mpip-mainz.mpg.de; weil@mpip-mainz.mpg.de
bGraduate School Materials Science in Mainz, Staudingerweg 9, 55128 Mainz, Germany
cDepartment of Dermatology, University Medical Center of the Johannes Gutenberg-University Mainz, Langenbeckstr. 1, 55131 Mainz, Germany
First published on 19th June 2020
Abstract
Amphiphilic polyphenylene dendrimers (PPDs) with distinct lipophilic and positively or negatively charged surface groups were adsorbed onto liposomes and their impact on protein adsorption in blood plasma was studied. The PPD corona reduced binding of specific opsonins and increased the adsorption of proteins controlling cellular uptake based on their surface patches.
The formation of a protein corona on the surface of nanoparticles is a critical factor that determines their biodistribution, cellular uptake pathways and immune response. There are many parameters that influence protein adsorption on nanocarriers such as their size, surface charges, hydrogen bonds and van der Waals interactions.1 Liposomes are among the most applied nanocarriers, but their biological fate is hard to control. A deeper understanding which surface groups facilitate binding of certain plasma proteins is still elusive. Polyethylene glycol surfaces usually reveal low protein adsorption and stealth-like properties.1 However, instead of just limiting protein binding, it would be very attractive to design surface coatings that only adsorb certain proteins from blood plasma and thus, enhance cellular uptake or affect biodistribution. To understand and control protein corona formation, novel coatings mimicking features of proteins are designed providing amphiphilic patches with low nanometre dimensions due to hydrophilic, hydrophobic and charged surface groups at defined locations. Dendrimers are monodisperse macromolecules with customizable surface groups and nanometre dimensions that are often considered as protein mimics.2 However, many aliphatic dendrimers exhibit backfolding of their dendritic branches. Thus, surface groups are not exposed exclusively at the surface resulting in less defined peripheral patterns.3 Widely used polycationic poly(amidoamine) (PAMAM) dendrimers interact with blood plasma proteins but they also reveal high cellular toxicities and trigger immune responses.4
Herein, we apply polyphenylene dendrimers (PPDs) as coatings for liposomal nanocarriers to assess the impact of amphiphilic surface groups on plasma protein binding. The PPDs consist of a semi-rigid polyphenylene scaffold with no backfolding of dendritic branches, alternating sulfonic acid and n-propyl groups, and they reveal cellular uptake and low toxicity.5 In blood plasma, they form a novel PPD-corona at the surface of adenovirus 5 preventing the endogenous blood coagulation factor X from binding.6 Liposomal nanocarriers coated with PPDs reveal a considerably altered protein corona in blood serum.7
Herein, PPDs with either positively or negatively charged amphiphilic or exclusively anionic surface groups were prepared and the impact on protein corona formation was studied. We envision controlling the fate of liposomal nanocarriers by the PPD surface in biological fluids (Fig. 1).
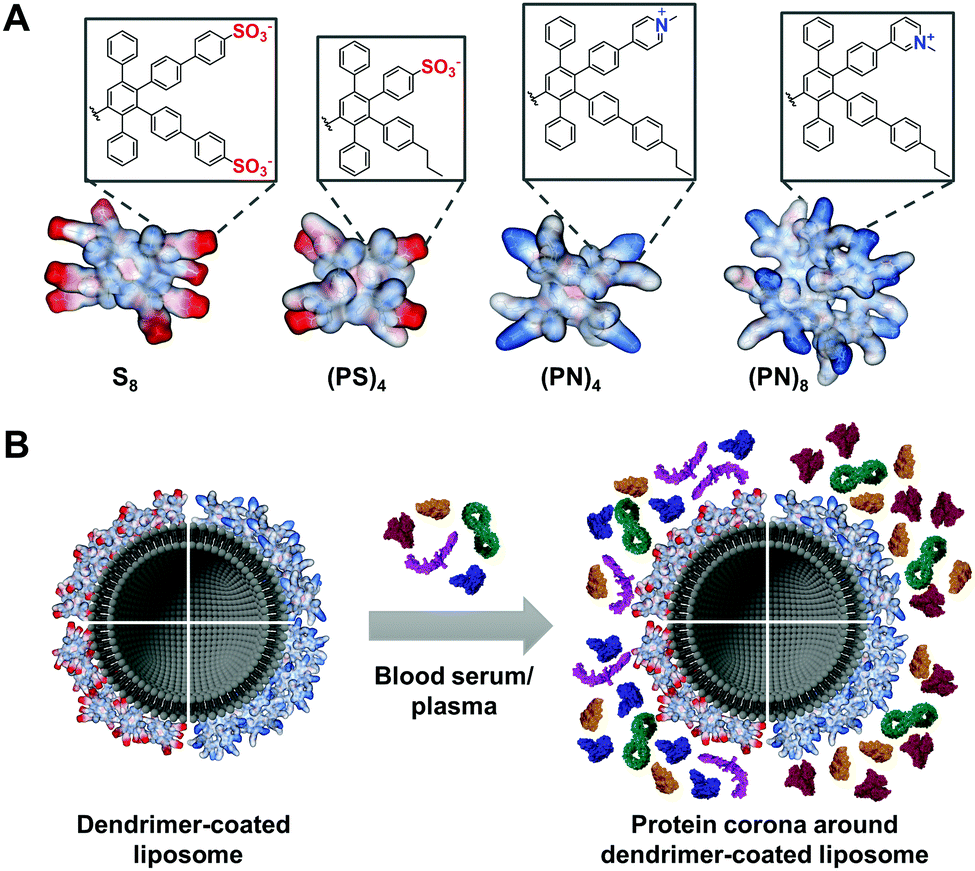 | ||
| Fig. 1 Protein corona on PPD-coated liposomes. (A) PPDs with various surface pattern consisting of sulfonic acid groups (S8), alternating sulfonic acid and n-propyl groups ((PS)4) or alternating pyridinium and n-propyl groups ((PN)4 and (PN)8), (P = n-propyl, S = sulfonate, N = pyridinium); (B) PPD-coating on liposomes alters the protein corona in blood serum and plasma. | ||
The synthesis of charged PPDs was performed according to the divergent growth approach (Scheme 1 and Fig. S2–S5, ESI†). Growth of the PPDs starts from an ethynylated core, which is reacted with tetraphenylcyclopentadienone (CP) building blocks in a [4+2] Diels–Alder cycloaddition. The CP determines the branching and the absolute surface pattern of the dendrimer.8
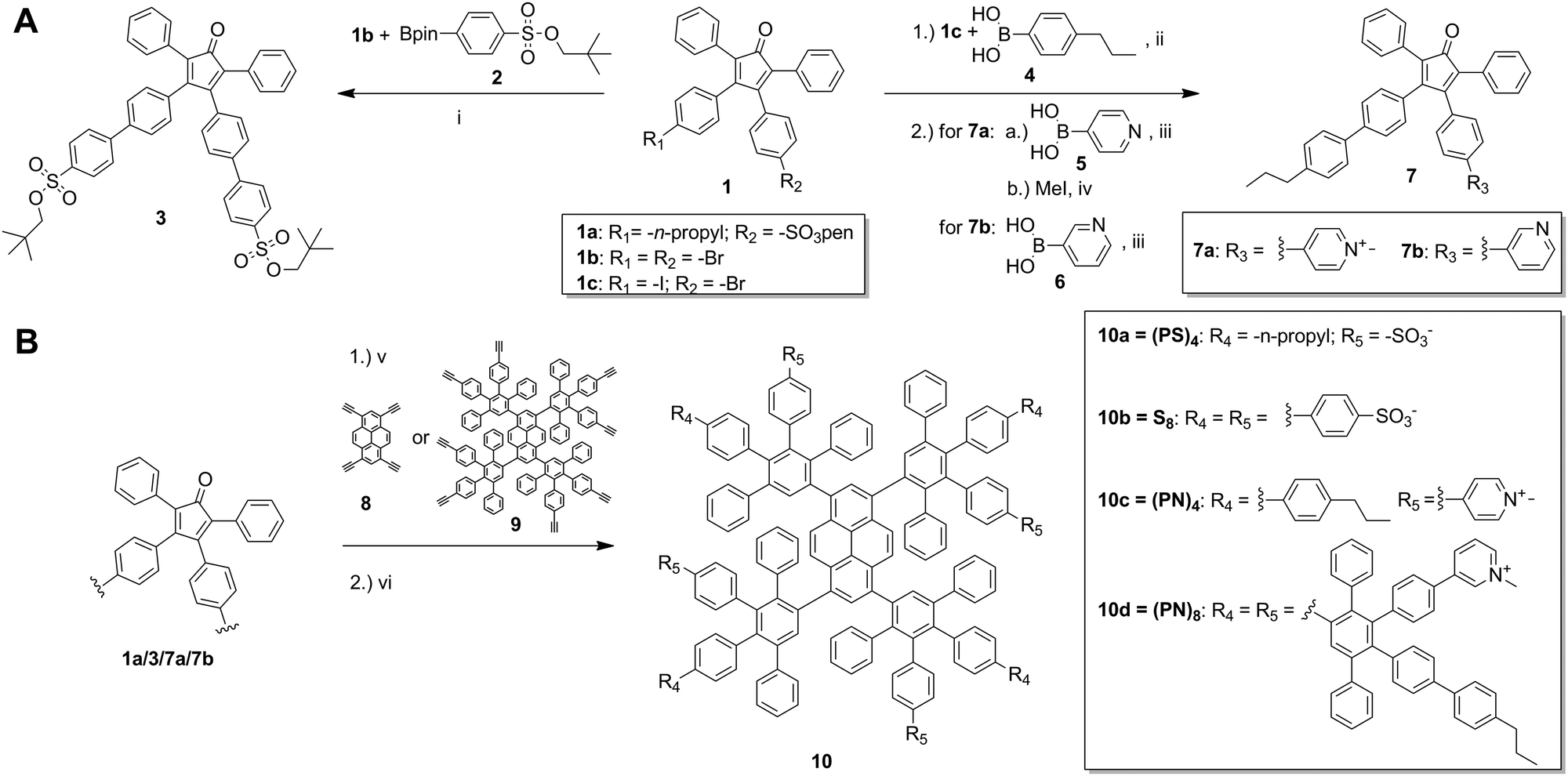 | ||
| Scheme 1 Synthesis of PPDs with different surface patterns. (A) Building block syntheses: (i) Pd(PPh3)4, K2CO3 (aq), 1,4-dioxane, 90 °C, 15 h, 34%; (ii) Pd(PPh3)4, K2CO3 (aq), 1,4-dioxane, 40 °C, 15 h; (iii) Pd(PPh3)4, K2CO3 (aq), 1,4-dioxane, 80 °C, 48 h, 50–60%; (iv) methanol, RT, 48 h, 70%; (B) dendrimer synthesis: (v) 1a/3: pyrene-core 8, o-xylene, 145 °C, 48 h, 27–45%; 7a: pyrene-core 8, DMSO, 140 °C, 3 d, 47%; 7b: first-generation dendrimer core 9, o-xylene, 160 °C, 3 d, 64%; (vi) deprotection sulfonic acid groups to obtain 10a, 10b: DMF, 180 °C, 48 h, 85–88%; methylation to obtain 10d: methyl iodide, methanol, RT, 24 h, 92%. | ||
Building block 1a with a sulfonate and n-propyl group was reported before7,9 and was directly used for the PPD growth whereas 1b (Fig. S3, ESI†) and 1c (Fig. S4, ESI†) were further modified with the desired surface functionalities by Suzuki coupling reactions (Scheme 1). For the anionic PPD with peripheral sulfonate groups, intermediate 39 with two neopentyl-protected sulfonic acid moieties was synthesized by C–C coupling of the pinacol boronic ester 2 to di-bromo-modified compound 1b. For cationic amphiphilic PPDs, CP 1c with an iodo- and bromo-substituent was synthesized based on modified protocols (Fig. S4, ESI†).10 Terminal surface groups were attached to 1c by Suzuki coupling of n-propyl functionalized phenyl derivative 4 and either meta- or para-substituted pyridine compounds 5 and 6 (Scheme 1). To achieve a positively charged pyridinium group on the dendrimer rim, the pyridine moieties were methylated. For the first-generation PPDs, methylation was performed prior to dendrimer synthesis (a priori) to obtain pyridinium modified CP 7a. When synthesizing a second-generation PPDs, the pyridine group was methylated after dendrimer growth, due to incomplete dendrimer formation with already cationic building blocks at higher generations. In addition, the meta-pyridinyl group of 7b facilitated the methylation on the dendrimer surface. All PPDs are built around the core of 1,3,6,8-tetraethynylpyrene (8). The cycloaddition to achieve both neopentyl-protected sulfonic acid-based dendrimers were performed at 145 °C in o-xylene for 48 h.5,9 After purification by silica gel column chromatography and recycling-gel permeation chromatography (GPC), the neopentyl protective groups of the sulfonic acids were thermally cleaved to obtain 10a and 10b.5,9 Deprotected PPDs were further purified via GPC in DMF. The positively charged amphiphilic PPD 10c was obtained by cycloaddition of pyrene core 8 and 7a in DMSO at 140 °C for 3 d. The product was precipitated and purified by dialysis. The second-generation PPD was synthesized from a first-generation pyrene-based PPD 9,5 which was reacted with pyridine modified CP 7b in o-xylene at 160 °C for 3 d. After purification by GPC, the product was methylated. Cationic amphiphilic PPD 10d was obtained by precipitation in diethyl ether. NMR spectroscopy and MALDI-TOF or ESI mass spectroscopy confirmed the structure of 10a–10d.
We coated liposomes and polystyrene nanoparticles with the PPDs and investigated protein corona formation in blood serum and plasma. We used similar liposome composition as reported previously,7 consisting of 2-dioleoyl-sn-glycero-3-phosphoethanolamine (DOPE), L-α-phosphatidylcholine (eggPC) and cholesterol (Chol) (PC![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :DOPE:Chol = 1:1:1) as well as amino-functionalized polystyrene nanoparticles (PS-NH2), which were prepared according to standard protocols.11 Liposomes or PS-NH2 were mixed with the dendrimers 10a (PS)4, 10b S8, 10c (PN)4 or 10d (PN)8 (with P = n-propyl, S = sulfonate and N = pyridinium). Coating of both nanocarriers was verified by a shift of ζ-potential values (Fig. S6, ESI†). Interestingly, only for (PS)4 and (PN)8 liposome coating was observed (Fig. S6B, ESI†). It has been reported that amphiphilic PPDs with alternating sulfonic acid (S) and n-propyl groups (P) bind to a lipid monolayer by electrostatic interactions.12 Since S8 did not interact with liposomes efficiently (Fig. S6B, ESI†), we postulate that the hydrophobic interactions between n-propyl groups and lipid tails is necessary to enhance binding to liposomes. In addition, second-generation dendrimer (PN)8 coated liposomes revealed protein binding to a higher extent than (PN)4 resulting in a positive ζ-potential. Sufficient PPD surface coverage of liposomes was only achieved for (PS)4 and (PN)8. To assess the impact of the n-propyl (P) group on the protein corona formation, we coated PS-NH2 nanoparticles with (PS)4 and S8. Due to the positive ζ-potential of amino-functionalized PS particles, adsorption of negatively charged PPDs is electrostatically driven. This was further supported by using positively charged (PN)4 and (PN)8, which did not show much binding to PS-NH2 (Fig. S6C, ESI†). Thus, either blood serum or blood plasma was added to the PPD-coated liposomes (lipo-dendrimer) or PS-NH2 nanoparticles. Protein adsorption was analysed quantitatively by Pierce Assay (Fig. S7, ESI†) and LC-MS/MS (Fig. 2 and Fig. S8, S9, ESI†). For the blood serum preparation, fibrinogen and other blood clotting factors were removed by centrifugation, whereas blood plasma contained all proteins including clotting factors. For plasma, clotting was prevented by the addition of citrate as anticoagulant. Using the Pierce Assay, we could further confirm the binding efficiency of dendrimers to the particle surfaces by comparing the protein adsorption levels to uncoated nanocarriers (Fig. S7B–E, ESI†). Furthermore, protein quantities increased for higher dendrimer concentrations, which did not largely affect the protein corona composition (Fig. S12–S14, ESI†). The heatmaps depict an overview of all blood proteins bound onto lipo-dendrimers (lipo-(PS)4 and lipo-(PN)8; Fig. 2A and Fig. S8, ESI†) as well as PS-dendrimers (PS–(PS)4 and PS–S8) (Fig. S9, ESI†) in blood plasma and serum. First, we observed similar changes in the adsorption of certain proteins to uncoated liposomes or lipo-dendrimers regardless of the surface charges of (PS)4 and (PN)8 (Fig. 2B). Both dendrimer surfaces lead to a significant reduction of the opsonins immunoglobulin γ-2 (Ig γ-2) and complement C3 levels in comparison to uncoated liposomes (Fig. 2C and D). Opsonins are recognized by immune cells, which are part of the mononuclear phagocytic system (MPS), and they mediate cellular uptake of nanocarriers into phagocytic cells.1 Thus, these opsonins enhance blood clearance and reduce the interaction with targeted cells. In contrast, we observed an enhancement in clusterin binding from uncoated to covered liposomes (Fig. 2E). Clusterin, also termed Apolipoprotein J, decreased unspecific cellular uptake of PEGylated nanocarriers in vitro and functioned as a dysopsonin for macrophages.13 In general, apolipoproteins bind to a higher extent to hydrophobic nanocarriers.14 Thus, we assume that the clusterin binding is related to the hydrophobic character of PPDs. Furthermore, fibrinogen was adsorbed on all lipo-dendrimers (Fig. 2F). Previously, the pre-coating of PS nanoparticles with IgG-depleted plasma furnished a high enrichment of fibrinogen and a reduced cellular uptake in macrophages.15 In addition, the dendrimer coating gave rise to accelerated vitronectin binding (Fig. 2G). These findings were consistent with our previous studies for dendrimers and dendrimer branches with higher density of sulfonic acid and n-propyl groups (Fig. S10, ESI†).7 Vitronectin was reported to mediate a selective uptake of lipoplexes into cancer cells with overexpressing αvβ3 vitronectin receptors.16 Fibrinogen and vitronectin binding occurred for PS–(PS)4 and PS–S8 (Fig. S11C and D, ESI†) emphasizing that binding of these proteins is attributable to the hydrophobic PPD scaffold.
:DOPE:Chol = 1:1:1) as well as amino-functionalized polystyrene nanoparticles (PS-NH2), which were prepared according to standard protocols.11 Liposomes or PS-NH2 were mixed with the dendrimers 10a (PS)4, 10b S8, 10c (PN)4 or 10d (PN)8 (with P = n-propyl, S = sulfonate and N = pyridinium). Coating of both nanocarriers was verified by a shift of ζ-potential values (Fig. S6, ESI†). Interestingly, only for (PS)4 and (PN)8 liposome coating was observed (Fig. S6B, ESI†). It has been reported that amphiphilic PPDs with alternating sulfonic acid (S) and n-propyl groups (P) bind to a lipid monolayer by electrostatic interactions.12 Since S8 did not interact with liposomes efficiently (Fig. S6B, ESI†), we postulate that the hydrophobic interactions between n-propyl groups and lipid tails is necessary to enhance binding to liposomes. In addition, second-generation dendrimer (PN)8 coated liposomes revealed protein binding to a higher extent than (PN)4 resulting in a positive ζ-potential. Sufficient PPD surface coverage of liposomes was only achieved for (PS)4 and (PN)8. To assess the impact of the n-propyl (P) group on the protein corona formation, we coated PS-NH2 nanoparticles with (PS)4 and S8. Due to the positive ζ-potential of amino-functionalized PS particles, adsorption of negatively charged PPDs is electrostatically driven. This was further supported by using positively charged (PN)4 and (PN)8, which did not show much binding to PS-NH2 (Fig. S6C, ESI†). Thus, either blood serum or blood plasma was added to the PPD-coated liposomes (lipo-dendrimer) or PS-NH2 nanoparticles. Protein adsorption was analysed quantitatively by Pierce Assay (Fig. S7, ESI†) and LC-MS/MS (Fig. 2 and Fig. S8, S9, ESI†). For the blood serum preparation, fibrinogen and other blood clotting factors were removed by centrifugation, whereas blood plasma contained all proteins including clotting factors. For plasma, clotting was prevented by the addition of citrate as anticoagulant. Using the Pierce Assay, we could further confirm the binding efficiency of dendrimers to the particle surfaces by comparing the protein adsorption levels to uncoated nanocarriers (Fig. S7B–E, ESI†). Furthermore, protein quantities increased for higher dendrimer concentrations, which did not largely affect the protein corona composition (Fig. S12–S14, ESI†). The heatmaps depict an overview of all blood proteins bound onto lipo-dendrimers (lipo-(PS)4 and lipo-(PN)8; Fig. 2A and Fig. S8, ESI†) as well as PS-dendrimers (PS–(PS)4 and PS–S8) (Fig. S9, ESI†) in blood plasma and serum. First, we observed similar changes in the adsorption of certain proteins to uncoated liposomes or lipo-dendrimers regardless of the surface charges of (PS)4 and (PN)8 (Fig. 2B). Both dendrimer surfaces lead to a significant reduction of the opsonins immunoglobulin γ-2 (Ig γ-2) and complement C3 levels in comparison to uncoated liposomes (Fig. 2C and D). Opsonins are recognized by immune cells, which are part of the mononuclear phagocytic system (MPS), and they mediate cellular uptake of nanocarriers into phagocytic cells.1 Thus, these opsonins enhance blood clearance and reduce the interaction with targeted cells. In contrast, we observed an enhancement in clusterin binding from uncoated to covered liposomes (Fig. 2E). Clusterin, also termed Apolipoprotein J, decreased unspecific cellular uptake of PEGylated nanocarriers in vitro and functioned as a dysopsonin for macrophages.13 In general, apolipoproteins bind to a higher extent to hydrophobic nanocarriers.14 Thus, we assume that the clusterin binding is related to the hydrophobic character of PPDs. Furthermore, fibrinogen was adsorbed on all lipo-dendrimers (Fig. 2F). Previously, the pre-coating of PS nanoparticles with IgG-depleted plasma furnished a high enrichment of fibrinogen and a reduced cellular uptake in macrophages.15 In addition, the dendrimer coating gave rise to accelerated vitronectin binding (Fig. 2G). These findings were consistent with our previous studies for dendrimers and dendrimer branches with higher density of sulfonic acid and n-propyl groups (Fig. S10, ESI†).7 Vitronectin was reported to mediate a selective uptake of lipoplexes into cancer cells with overexpressing αvβ3 vitronectin receptors.16 Fibrinogen and vitronectin binding occurred for PS–(PS)4 and PS–S8 (Fig. S11C and D, ESI†) emphasizing that binding of these proteins is attributable to the hydrophobic PPD scaffold.
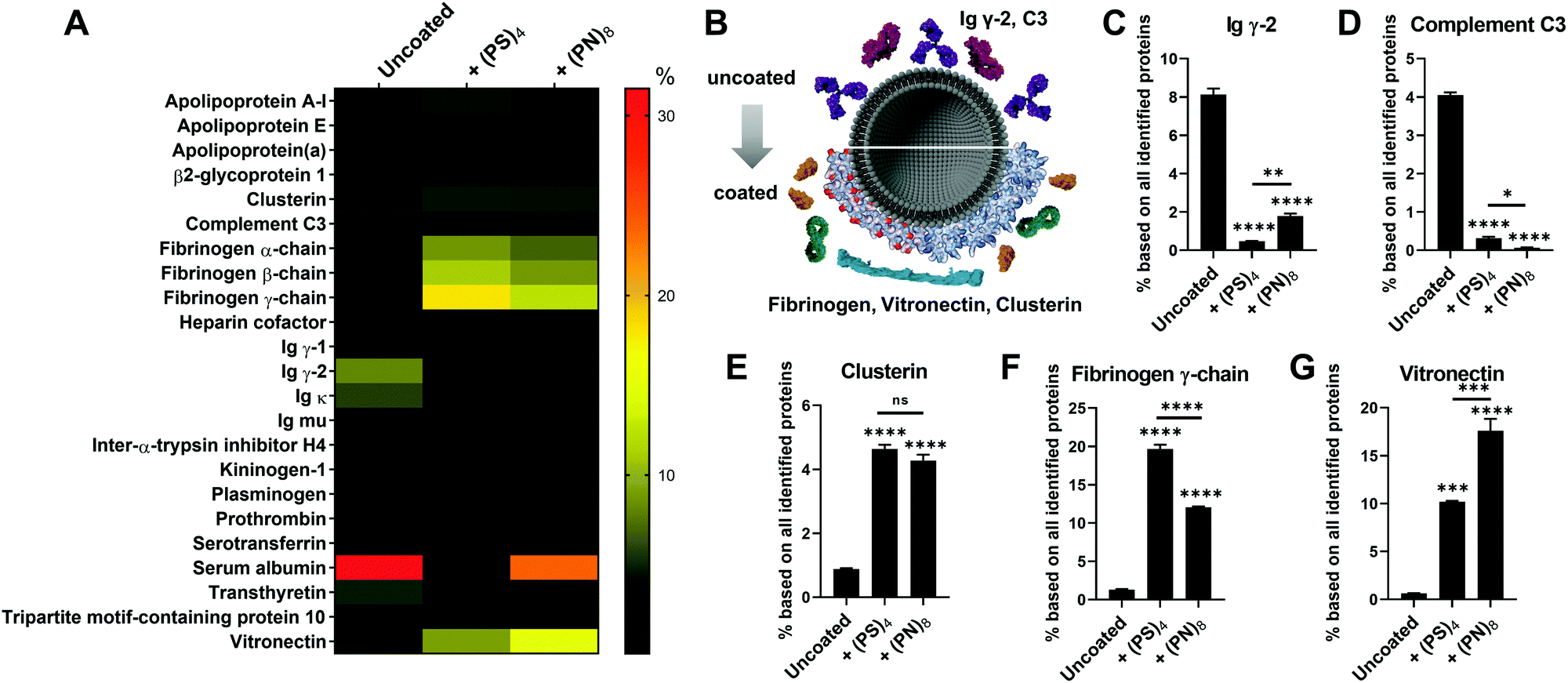 | ||
| Fig. 2 Comparison of adsorbed proteins between uncoated liposomes and lipo-dendrimers (lipo-(PS)4 and lipo-(PN)8) in human plasma. (A) Heatmap of adsorbed proteins to lipo-(PS)4 and lipo-(PN)8 in human plasma. Each protein amount is given in % relative to the total amount of all identified proteins. A list of all identified proteins is provided in Table S1 (ESI†). (B) Coating of liposomes lead to a different protein corona: reduction of (C) Ig γ-2 adsorption and (C) complement C3 and enhancement of (E) clusterin, (F) fibrinogen and (G) vitronectin (n = minimum 2; one-way ANOVA; not significant (ns) for p > 0.05, * for p ≤ 0.05, ** for p ≤ 0.01, *** for p ≤ 0.001, **** for p ≤ 0.0001). | ||
Second, we also observed remarkable differences in the protein corona that might be caused by the surface charges (Fig. 3A). Binding of serum albumin (HSA) for lipo-(PN)8 was enhanced whereas lipo-(PS)4 only showed a very low HSA adsorption (Fig. 3B). HSA is reported to serve as dysopsonin leading to higher blood circulation times.17 PS–(PS)4 and PS–S8 also displayed a reduction of HSA in the protein corona compared to uncoated nanoparticles (Fig. S11B, ESI†). This suggests that the positively charged pyridinium group is involved in the interaction of lipo-(PN)8 with HSA. In contrast, inter-α-trypsin inhibitor H4 was increased for all negatively charged liposomes and nanoparticles (lipo-(PS)4 (Fig. 3C), PS–(PS)4 and PS–S8 (Fig. S11E, ESI†)), which is known to interact with the highly negatively charged hyaluronic acid.18 Additionally, lipo-(PS)4 adsorbed β2-glycoprotein 1 also known as Apolipoprotein H (ApoH, Fig. 3D), which is consistent with our previous findings.7 ApoH binds to negatively charged phospholipids and mediates cellular uptake into mesenchymal stem cells.19 For PS–S8, we observed an even higher ApoH adsorption, suggesting that this interaction could be favoured by the peripheral sulfonates (Fig. S11F, ESI†).
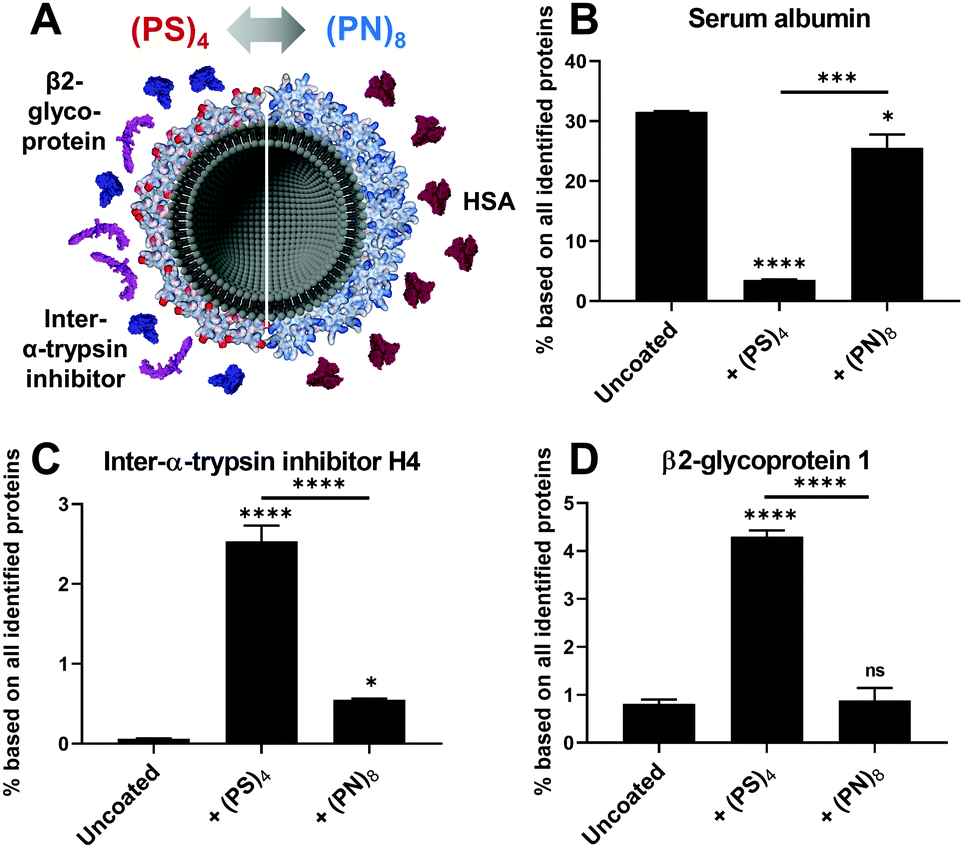 | ||
| Fig. 3 Comparison of adsorbed proteins on lipo-(PS)4 and lipo-(PN)8). (A) Differences in the protein corona of lipo-(PS)4 and lipo-(PN)8) summarizing the binding results of (B) HSA, (C) inter-α-trypsin-inhibitor H4 as well as (D) β2-glycoprotein 1 (n = minimum 2; one-way ANOVA; ns for p > 0.05, * for p ≤ 0.05, *** for p ≤ 0.001, **** for p ≤ 0.0001). | ||
In conclusion, we synthesized PPDs with different surface patterns that offer amphiphilicity, varying charges and shape persistence resulting in distinct surface structures with nanoscale perfection. We could show that surface charges and hydrophobicity of PPDs alter the protein corona on liposomes. For all PPDs, a reduction of opsonization proteins and enhancement of proteins, which might control selective cellular uptake, were observed. Thus, we demonstrated that the protein corona of nanoparticles is modulated through PPD coating, which opens new avenues to control their biodistribution in vivo.
We thank the Volkswagen Foundation (Project No. 88396), the German Research Foundation (Project No. 213555243, SFB 1066) for financial support and P. Schiel, M. Gai and K. Klein for technical support and S. Schuhmacher for graphical assistance. Open Access funding provided by the Max Planck Society.
Conflicts of interest
There are no conflicts to declare.Notes and references
- S. Schöttler, K. Landfester and V. Mailänder, Angew. Chem., Int. Ed., 2016, 55, 8806 CrossRef PubMed.
- R. M. Kannan, E. Nance, S. Kannan and D. A. Tomalia, J. Intern. Med., 2014, 276, 579 CrossRef CAS PubMed.
- P. K. Maiti, T. Çaǧın, S.-T. Lin and W. A. Goddard, Macromolecules, 2005, 38, 979 CrossRef CAS.
- A. Åkesson, M. Cárdenas, G. Elia, M. P. Monopoli and K. A. Dawson, RSC Adv., 2012, 2, 11245 RSC.
- R. Stangenberg, Y. Wu, J. Hedrich, D. Kurzbach, D. Wehner, G. Weidinger, S. L. Kuan, M. I. Jansen, F. Jelezko, H. J. Luhmann, D. Hinderberger, T. Weil and K. Müllen, Adv. Healthcare Mater., 2015, 4, 377 CrossRef CAS PubMed.
- Y. Wu, L. Li, L. Frank, J. Wagner, P. Andreozzi, B. Hammer, M. D’Alicarnasso, M. Pelliccia, W. Liu, S. Chakrabortty, S. Krol, J. Simon, K. Landfester, S. L. Kuan, F. Stellacci, K. Müllen, F. Kreppel and T. Weil, ACS Nano, 2019, 13, 8749 CrossRef CAS PubMed.
- J. Wagner, L. Li, J. Simon, L. Krutzke, K. Landfester, V. Mailänder, K. Müllen, D. Y. W. Ng, Y. Wu and T. Weil, Angew. Chem., Int. Ed., 2020, 59, 5712 CrossRef CAS PubMed.
- U. M. Wiesler, A. J. Berresheim, F. Morgenroth, G. Lieser and K. Müllen, Macromolecules, 2001, 34, 187 CrossRef CAS.
- R. Stangenberg, I. Saeed, S. L. Kuan, M. Baumgarten, T. Weil, M. Klapper and K. Müllen, Macromol. Rapid Commun., 2014, 35, 152 CrossRef CAS PubMed.
- G. Zhang, M. Auer-Berger, D. W. Gehrig, P. W. M. Blom, M. Baumgarten, D. Schollmeyer, E. J. W. List-Kratochvil and K. Müllen, Molecules, 2016, 21, 1400 CrossRef PubMed.
- M. Gai, J. Simon, I. Lieberwirth, V. Mailänder, S. Morsbach and K. Landfester, Polym. Chem., 2020, 11, 527 RSC; M. Kokkinopoulou, J. Simon, K. Landfester, V. Mailänder and I. Lieberwirth, Nanoscale, 2017, 9, 8858 RSC.
- M. Okuno, M. Mezger, R. Stangenberg, M. Baumgarten, K. Müllen, M. Bonn and E. H. G. Backus, Langmuir, 2015, 31, 1980 CrossRef CAS PubMed.
- S. Schöttler, G. Becker, S. Winzen, T. Steinbach, K. Mohr, K. Landfester, V. Mailänder and F. R. Wurm, Nat. Nanotechnol., 2016, 11, 372 CrossRef PubMed.
- A. Gessner, R. Waicz, A. Lieske, B. R. Paulke, K. Mäder and R. H. Müller, Int. J. Pharm., 2000, 196, 245 CrossRef CAS.
- J. Simon, L. K. Müller, M. Kokkinopoulou, I. Lieberwirth, S. Morsbach, K. Landfester and V. Mailänder, Nanoscale, 2018, 10, 10731 RSC.
- G. Caracciolo, F. Cardarelli, D. Pozzi, F. Salomone, G. Maccari, G. Bardi, A. L. Capriotti, C. Cavaliere, M. Papi and A. Laganà, ACS Appl. Mater. Interfaces, 2013, 5, 13171 CrossRef CAS PubMed.
- G. Caracciolo, S. Palchetti, V. Colapicchioni, L. Digiacomo, D. Pozzi, A. L. Capriotti, G. La Barbera and A. Laganà, Langmuir, 2015, 31, 10764 CrossRef CAS PubMed.
- L. Chen, S. J. Mao, L. R. McLean, R. W. Powers and W. J. Larsen, J. Biol. Chem., 1994, 269, 28282 CAS.
- S. Ritz, S. Schöttler, N. Kotman, G. Baier, A. Musyanovych, J. Kuharev, K. Landfester, H. Schild, O. Jahn, S. Tenzer and V. Mailänder, Biomacromolecules, 2015, 16, 1311 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0cc02486d |
| ‡ J. W. and M. D. contributed equally. |
| This journal is © The Royal Society of Chemistry 2020 |