
An N,N′-diethylformamide solvent-induced conversion cascade within a metal–organic framework single crystal†
Wen-Hua
Zhang  *a
and
David J.
Young
b
*a
and
David J.
Young
b
*a
and
David J.
Young
b
Abstract
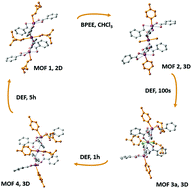
Crystals of a two-dimensional (2D) metal–organic framework (MOF) [Cd3(BTB)2(DEF)4]·2(DEF)0.5 (1; BTB = benzene-1,3,5-tribenzolate; DEF = N,N′-diethylformamide) immersed in a solution of trans-1,2-bis(4-pyridyl)ethylene (BPEE) yields an interpenetrated three-dimensional (3D) MOF of [Cd3(BTB)2(BPEE)(H2O)2]·(BPEE)·xSol (2). Crystals of MOF 2, in turn, undergo a cascade conversion when immersed in DEF, yielding [Cd3(BTB)2(BPEE)1.8(DEF)0.9(H2O)0.8]·xSol (3a) over 100 seconds and [Cd3(BTB)2(BPEE)2(DEF)2]·xSol (4) after one hour, before finally shuttling back to MOF 1 after six hours.
- This article is part of the themed collection: SU 120: Celebrating 120 Years of Soochow University
 Please wait while we load your content...
Please wait while we load your content...

