
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Chemical synthesis of human trefoil factor 1 (TFF1) and its homodimer provides novel insights into their mechanisms of action†
Nayara
Braga Emidio
 a,
Hayeon
Baik
b,
David
Lee
a,
René
Stürmer
c,
Jörn
Heuer
c,
Alysha G.
Elliott
a,
Mark A. T.
Blaskovich
a,
Katharina
Haupenthal
c,
Nicole
Tegtmeyer
d,
Werner
Hoffmann
c,
Christina I.
Schroeder
ae and
Markus
Muttenthaler
*ab
a,
Hayeon
Baik
b,
David
Lee
a,
René
Stürmer
c,
Jörn
Heuer
c,
Alysha G.
Elliott
a,
Mark A. T.
Blaskovich
a,
Katharina
Haupenthal
c,
Nicole
Tegtmeyer
d,
Werner
Hoffmann
c,
Christina I.
Schroeder
ae and
Markus
Muttenthaler
*ab
aInstitute for Molecular Bioscience, The University of Queensland, St Lucia, Brisbane, Queensland 4072, Australia
bInstitute of Biological Chemistry, Faculty of Chemistry, University of Vienna, Währingerstr. 38, Vienna, 1090, Austria. E-mail: markus.muttenthaler@univie.ac.at
cInstitute of Molecular Biology and Medicinal Chemistry, Otto-von-Guericke-University Magdeburg, Leipziger Str. 44, 39120 Magdeburg, Germany
dDivision of Microbiology, Department of Biology, Friedrich-Alexander-University, Erlangen-Nürnberg Staudtstr. 5, 91058 Erlangen, Germany
eNational Cancer Institute, National Institutes of Health, Frederick, MD 21702, USA
First published on 30th April 2020
Abstract
TFF1 is a key peptide for gastrointestinal protection and repair. Its molecular mechanism of action remains poorly understood with synthetic intractability a recognised bottleneck. Here we describe the synthesis of TFF1 and its homodimer and their interactions with mucins and Helicobacter pylori. Synthetic access to TFF1 is an important milestone for probe and therapeutic development.
TFF1 is a member of the trefoil factor family (TFF) of gastrointestinal peptides, well-known for their role in protecting and repairing the gastrointestinal tract.1–3 A substantial body of evidence supports TFF1's role in gastric protection.1–5 TFF1 is typically expressed in gastric surface mucous cells3 and TFF1 knockout (−/−) mice spontaneously develop antropyloric adenoma with about 30% progressing to carcinomas, establishing TFF1 as a tumour suppressor.6,7 By contrast, transgenic mice overexpressing TFF1 display increased resistance to gastrointestinal damage8 and TFF1 is ectopically expressed in an in vivo model of acetic acid-induced colitis.9 Administration of TFF1 has additional protective and healing effects in animal models of gastrointestinal injury.10–12 Consequently, TFF1 has therapeutic potential for gastrointestinal disorders including inflammatory bowel diseases (IBD) and non-steroidal anti-inflammatory drug (NSAID)-induced gastritis.13–16
TFF1 is a 60 residue peptide with three disulfide bonds (CysI–V, CysII–IV, CysIII–VI) resulting in a highly structured and conserved three-loop containing TFF domain.1,17 A free cysteine residue (CysVII) is located outside the TFF domain and enables homo- and heterodimerisation via an intermolecular disulfide bond (e.g. TFF1–gastrokine 2).5,18,19 The molecular mechanisms through which TFF1 protects the gastrointestinal mucosa are not fully understood,10,11 but could involve enhancement of cell migration due to its motogenic20 and anti-apoptotic21 effects (‘restitution’),22 a scavenger function for extracellular reactive oxygen/nitrogen species,5 and/or mucin interactions.3,5 Lack of synthetic access to TFF1 and associated probes is the main bottleneck hindering progress on identification of its mode of action on a molecular level. Recombinant expression and purification of TFF1 is challenging,18,23 and synthesis of TFF1 monomer was attempted in the past, but no correctly folded TFF1 was obtained nor characterised.24 Reliable access to TFF1 peptides through chemical synthesis would facilitate large scale production, library design for structure–activity relationship (SAR) studies and site-specific chemical modifications for molecular probes, setting the stage for therapeutic development. We thus set out to develop a synthetic strategy for the efficient production of bioactive TFF1 and its analogues.
Attempts to directly assemble TFF1 via solid phase peptide synthesis (SPPS) were unsuccessful and prompted us to switch to a two-fragment ligation strategy (Fig. 1). TFF1 was split between Gly31 and Cys32 which was chosen based on fragment size and favoured kinetics for native chemical ligation (NCL) (ligation proceeds faster at sterically less hindered thioester positions).25,26 Traditionally, thioester fragments are synthesised by tert-butyloxycarbonyl (Boc)-SPPS because the thioester is not stable under the basic conditions and nucleophiles required for Nα-Fmoc deprotection.27 Attempts to synthesise the N-terminal segment containing the thioester by manual Boc-SPPS were however unsuccessful, resulting in poor crude quality and low yield. Thus, a Fmoc-SPPS compatible strategy that uses a C-terminal hydrazide as a thioester surrogate was pursued (Fig. 1).28 TFF11–31 was produced with a C-terminal hydrazide that was converted to a thioester prior to ligation with TFF132–60 to form full-length reduced TFF11–60 (Fig. 2A and B). TFF11–60 was then folded at pH 8.5 (50 μM) for 48 hours, forming three disulfide bonds. TFF1 displayed a two-peak analytical HPLC profile with identical mass (Fig. 2C). Both peaks were collected and each re-run, yielding the same two-peak profile, confirming a single molecular entity. Such multi-peak conformational HPLC peak profiles are not uncommon with peptides and proteins29–32 and have also been observed with TFF3.33
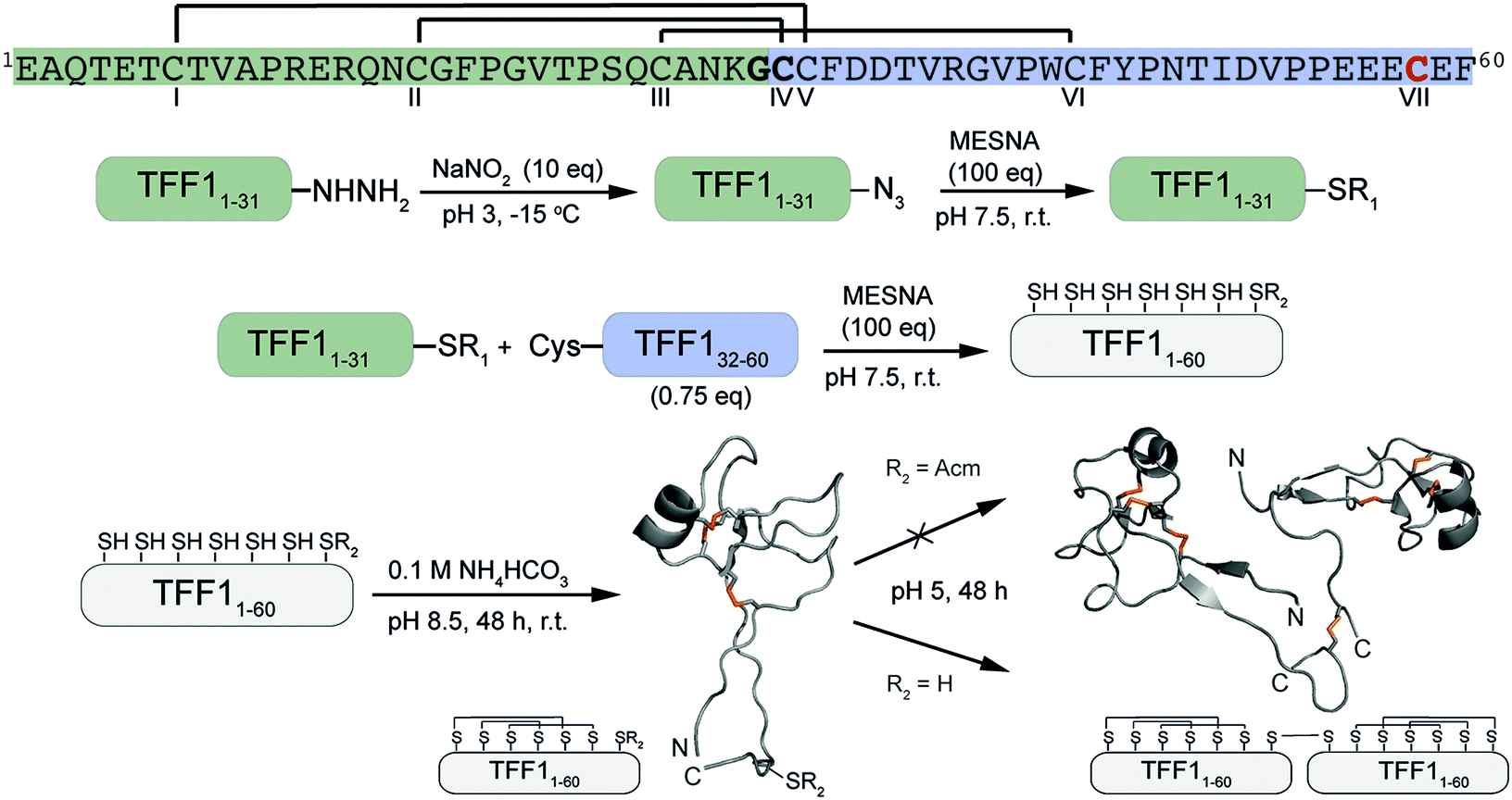 | ||
| Fig. 1 Synthetic strategy of the assembly of TFF1 monomer and homodimer. TFF1 sequence highlighted in green represents the N-terminal fragment and in blue the C-terminal fragment used for native chemical ligation. Residues in bold highlight the ligation site. CVII (red) enables dimerisation. Disulfide connectivity is indicated with black lines. NMR structures used for this scheme: TFF1 monomer PDB: 1PS2; TFF1 homodimer PDB: 1HI7. Disulfide bonds are shown in orange. | ||
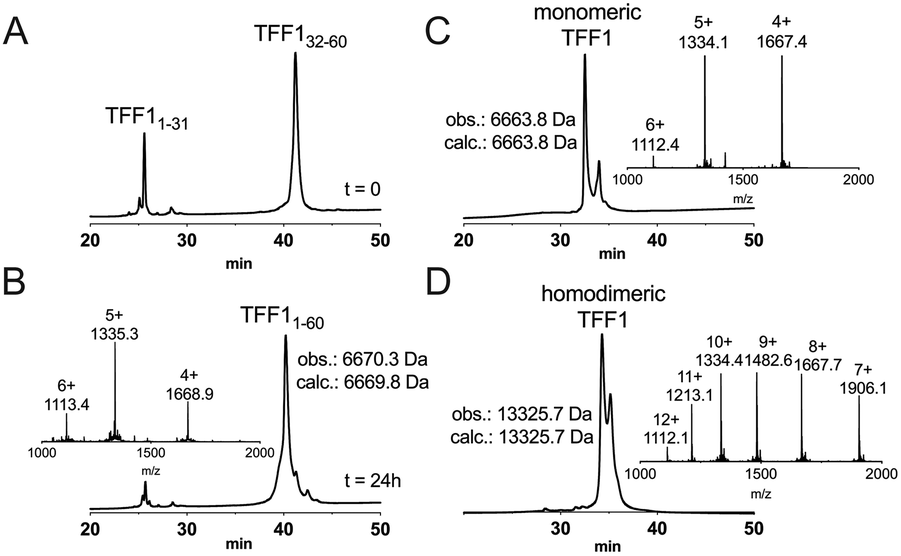 | ||
| Fig. 2 Chemical synthesis of TFF1 and TFF1 homodimer. (A) Analytical C18-RP-HPLC trace of NCL at 0 h. NCL occurred after the hydrazide fragment was converted into a thioester peptide through in situ activation and thiolysis. (B) Analytical C18-RP-HPLC and MS traces of NCL at 24 h. (C) Analytical C3-RP-HPLC and MS traces of folded TFF1. Reduced full-length TFF1 was oxidised in 0.1 M NH4HCO3, pH 8.5 for 48 h. (D) Analytical C3-RP-HPLC and MS traces of folded TFF1 homodimer. TFF1 was dimerised over 48 h in water. | ||
Homodimerisation of TFF1 was achieved through disulfide bond formation in water at high concentration (1.5 mM; pH ∼5, 48 h). TFF1 homodimer also displayed a two-peak HPLC profile (Fig. 2D) with identical mass. Since TFF1 dimerises slowly in water, we also synthesised TFF1 with CysVII protected (TFF1(C58Acm)) to ensure proper differentiation between TFF1 monomer and homodimer for our functional studies (Fig. S1, ESI†) and to have the option to unprotect CysVII when needed for dimer formation or conjugation to reporter tags. In previous studies with recombinant TFF1, undesired homodimerisation was achieved through a CysVII to Ser replacement.20
NMR spectroscopy confirmed the correct fold of both TFF1 and TFF1 homodimer. Hα chemical shifts were assigned using total correlated spectroscopy (TOCSY) and nuclear Overhauser effect spectroscopy (NOESY) and compared to the chemical shifts of recombinant TFF1 (BMRB: 4933)34 and TFF1 homodimer (BMRB: 4930).23 Synthetic and recombinant TFF1 analogues displayed good overlap of the secondary Hα chemical shifts (deviation of the Hα chemical shifts from random coil values35) confirming the same overall fold and 3D structure (Fig. 3). Circular dichroism (CD) experiments confirmed the presence of an α-helix characterised by negative bands at 208 nm and 222 nm and a positive band at 193 nm (Fig. S2, ESI†), corresponding well with the presence of negative secondary Hα chemical shifts indicative of an α-helix between position Pro24–Lys30 in loop 2 (Fig. 3).23,34
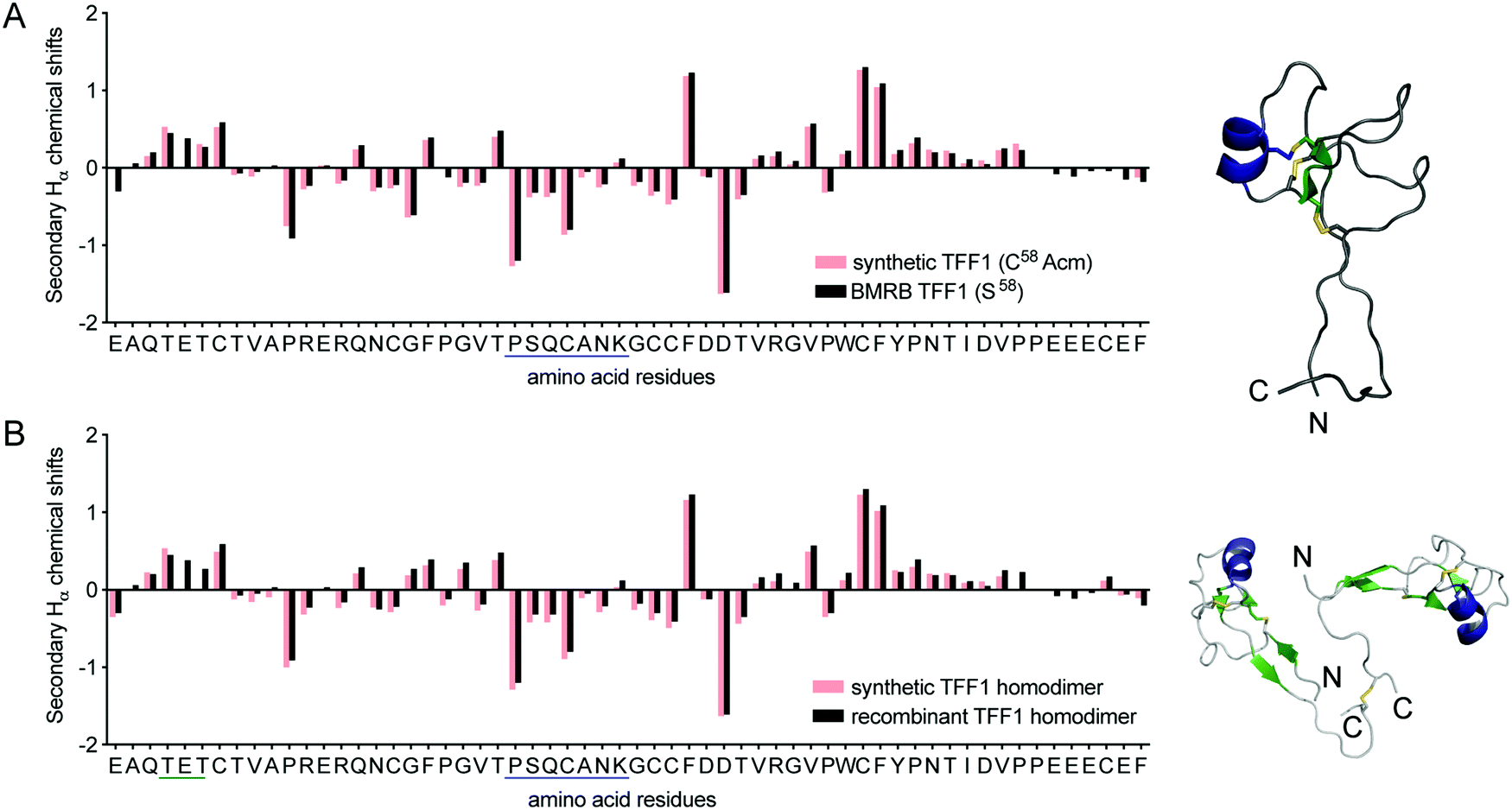 | ||
| Fig. 3 Structural secondary Hα chemical shift comparison of synthetic and recombinant TFF1 by NMR. (A) Comparison of synthetic TFF1(C58Acm) and recombinant TFF1(S58) (BMRB: 4933).34 (B) Comparison of synthetic TFF1 and recombinant TFF1 homodimer (BMRB: 4930).23 A putative α-helix is highlighted in blue in the sequence and NMR structure (TFF1 monomer PDB: 1PS2; TFF1 homodimer PDB: 1HI7). | ||
Interaction of TFF1 (C58Acm) and TFF1 homodimer with carbohydrates was tested in different mucin binding assays since TFF1 has been associated with lectin activities.36–39 Both peptides were labelled with 125I40 and then used in binding studies against purified mucin fractions from the stomach of human,41 pig40 and frog42 (Xenopus laevis) (Fig. 4).
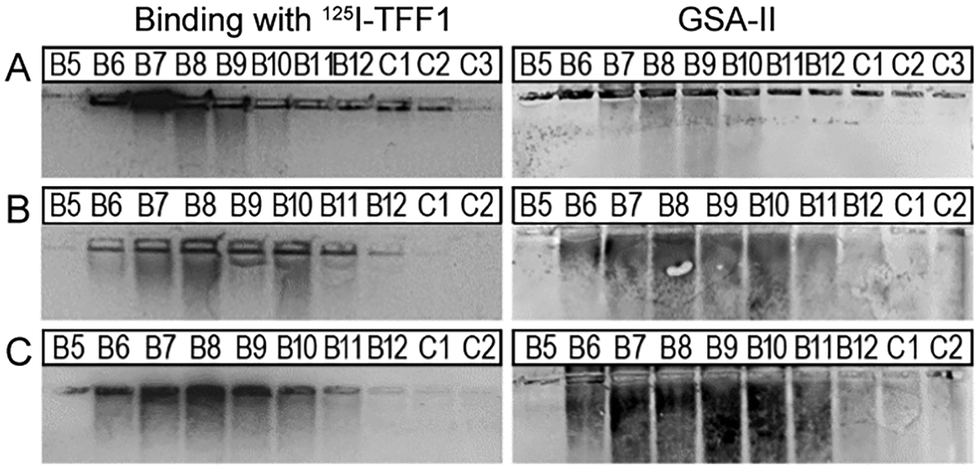 | ||
| Fig. 4 Binding studies of 125I-TFF1 homodimer with gastric mucins. Autoradiography was obtained after incubating 125I-TFF1 homodimer with different fractions purified (B5–C2/C3) by size exclusion chromatography from (A) human gastric mucins,41 (B) reduced porcine gastric mucins,40 and (C) gastric mucins from X. laevis.42 These fractions were separated by non-denaturing agarose gel electrophoresis which was directly blotted on a nitrocellulose membrane. Reactivity with GSA-II, a lectin from Griffonia simplicifolia, confirms the presence of mucins since it recognises αGlcNAc (α1,4-linked N-acetylglucosamine), a conserved carbohydrate motif in gastric mucins. | ||
In these overlay assays, only TFF1 homodimer bound to these mucins. Attempts to bind TFF1 (C58Acm) to mucin preparations failed. This indicates that dimerisation of TFF1 is essential for mucin interaction. This is in agreement with other observations that have also highlighted dimerisation as a critical factor for the protective function of TFF1.43,44125I-TFF1 homodimer also bound to a reduced porcine mucin preparation (boiled with 1% β-mercaptoethanol) (Fig. 4B), suggesting lectin-like binding to the carbohydrate moieties and not to its protein structure.36–38 Binding to frog mucins demonstrated that the gastric mucin sugar epitope recognised by TFF1 is evolutionary conserved (Fig. 4C). GSA-II, a lectin from Griffonia simplicifolia that recognises αGlcNAc (α1,4-linked N-acetylglucosamine; a conserved carbohydrate motif in gastric mucins), confirmed the presence of mucins. As a positive control and proof of synthetic TFF1 homodimer lectin activity, binding with cell lysates of H. pylori was tested (Fig. 5). A clear positive signal with the lipopolysaccharide was obtained with the H. pylori wild type in agreement with previous reports.36–39 In contrast, no binding was observed with a H. pylori mutant lacking seduheptulose 7-phosphate isomerase (P12ΔHP0857) which results in a truncated lipopolysaccharide core oligosaccharide.45
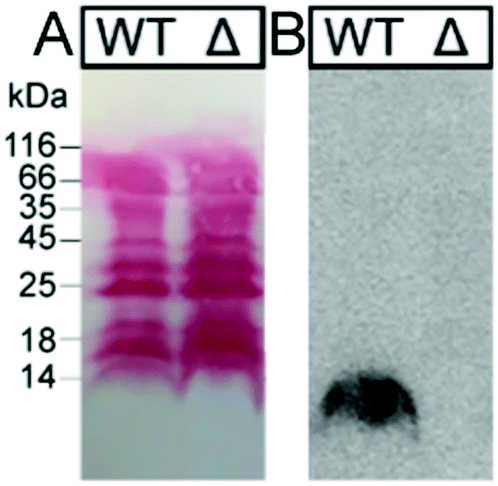 | ||
| Fig. 5 Binding studies of H. pylori extracts with 125I-TFF1 homodimer. H. pylori cell lysates (WT: wild type P12; Δ: mutant P12ΔHP0857) were run on a 15% SDS-PAGE and blotted on a nitrocellulose membrane. (A) Protein staining with Ponceau S showed that the mutation did not alter total protein expression. (B) Incubation of 125I-TFF1 with H. pylori cell lysates and autoradiography showed binding to WT but no binding to H. pylori mutant with a truncated lipopolysaccharide core oligosaccharide. | ||
TFF1 has also been implicated in cell migration and proliferation.3,20,21 However, independent confirmation, target identification and in-depth pharmacology remain sparse for these studies.1 With multi-milligrams of TFF1 at hand, our goal was to validate some of these studies in well-controlled experiments. We carried out cell migration and proliferation assays with HT-29 (human colorectal adenocarcinoma) cells using a state-of-the-art IncuCyte® system. In these studies, neither TFF1 nor TFF1 homodimer had any effect on cell migration or proliferation at concentrations of 0.1–10 μM (Fig. S3, ESI†). We also performed toxicity assays with TFF1 and its homodimer. Neither peptide displayed any cytotoxic or haemolytic effects at concentrations up to 25 μM (Fig. S4, ESI†).
In conclusion, we developed a synthetic strategy for TFF1, its homodimer and analogues. TFF1 homodimer bound to gastric mucins supporting the consensus that TFF1 elicits its protective action in the gut via lectin-like mucin cross-linking. We could not reproduce TFF1 activity in well-controlled HT-29 cell migration or proliferation assays (see ESI† for further discussion). Chemical access to TFF1 and its analogues represents an important milestone for the TFF field since it enables the production of homogenous material in addition to regioselective incorporation of chemical modifications that will facilitate molecular probe development for more in-depth mechanistic and target validation studies as well as therapeutic development. This in turn will advance our understanding of TFF1's physiology and therapeutic potential.
M. M. was supported by the European Research Council under the European Union's Horizon 2020 Research and Innovation Program (714366) and by the Australian Research Council (DE150100784, DP190101667). C. I. S. was an ARC Future Fellow (FT160100055). N. B. E. was supported by the University of Queensland International Postgraduate Scholarship. We thank Janet Reid and CO-ADD (the Community for Open Antimicrobial Drug Discovery), funded by the Wellcome Trust (Strategic Award 104797/Z/14/Z) and the University of Queensland, for the cytotoxicity and haemolysis assays. We thank Prof. S. Backert (FAU Erlangen-Nürnberg) and Prof. Paul Alewood (The University of Queensland) for their support of this work.
Conflicts of interest
There are no conflicts to declare.Notes and references
- N. Braga Emidio, W. Hoffmann, S. M. Brierley and M. Muttenthaler, Trends Biochem. Sci., 2019, 44, 387–390 CrossRef CAS PubMed.
- D. Taupin and D. K. Podolsky, Nat. Rev. Mol. Cell Biol., 2003, 4, 721–732 CrossRef CAS PubMed.
- S. Ribieras, C. Tomasetto and M. C. Rio, Biochim. Biophys. Acta, Rev. Cancer, 1998, 1378, F61–F77 CrossRef CAS.
- E. B. Znalesniak, F. Salm and W. Hoffmann, Int. J. Mol. Sci., 2020, 21, 644–659 CrossRef PubMed.
- J. Heuer, F. Heuer, R. Stürmer, S. Harder, H. Schlüter, N. Braga Emidio, M. Muttenthaler, D. Jechorek, F. Meyer and W. Hoffmann, Int. J. Mol. Sci., 2020, 21, 2508–2526 CrossRef PubMed.
- O. Lefebvre, M. P. Chenard, R. Masson, J. Linares, A. Dierich, M. LeMeur, C. Wendling, C. Tomasetto, P. Chambon and M. C. Rio, Science, 1996, 274, 259–262 CrossRef CAS PubMed.
- C. Tomasetto and M. C. Rio, Cell. Mol. Life Sci., 2005, 62, 2916–2920 CrossRef CAS PubMed.
- R. J. Playford, T. Marchbank, R. A. Goodlad, R. A. Chinery, R. Poulsom, A. M. Hanby and N. A. Wright, Proc. Natl. Acad. Sci. U. S. A., 1996, 93, 2137–2142 CrossRef CAS PubMed.
- H. Itoh, M. Tomita, H. Uchino, T. Kobayashi, H. Kataoka, R. Sekiya and Y. Nawa, Biochem. J., 1996, 318, 939–944 CrossRef CAS PubMed.
- A. J. FitzGerald, M. Pu, T. Marchbank, B. R. Westley, F. E. B. May, J. Boyle, M. Yadollahi-Farsani, S. Ghosh and R. J. Playford, Peptides, 2004, 25, 793–801 CrossRef CAS PubMed.
- K. Vandenbroucke, W. Hans, J. Van Huysse, S. Neirynck, P. Demetter, E. Remaut, P. Rottiers and L. Steidler, Gastroenterology, 2004, 127, 502–513 CrossRef CAS PubMed.
- S. Caluwaerts, K. Vandenbroucke, L. Steidler, S. Neirynck, P. Vanhoenacker, S. Corveleyn, B. Watkins, S. Sonis, B. Coulie and P. Rottiers, Oral Oncol., 2010, 46, 564–570 CrossRef CAS PubMed.
- N. A. Wright, Gut, 2001, 48, 293–294 CrossRef CAS PubMed.
- C. Tomasetto, R. Masson, J. L. Linares, C. Wendling, O. Lefebvre, M. P. Chenard and M. C. Rio, Gastroenterology, 2000, 118, 70–80 CrossRef CAS.
- J. L. Newton, A. Allen, B. R. Westley and F. E. B. May, Gut, 2000, 46, 312–320 CrossRef CAS PubMed.
- C. Dunne, J. Naughton, G. Duggan, C. Loughrey, M. Kilcoyne, L. Joshi, S. Carrington, H. Earley, S. Backert, C. R. Masselot, F. E. B. May and M. Clyne, Microorganisms, 2018, 6, 44–58 CrossRef PubMed.
- L. Thim and F. E. B. May, Cell. Mol. Life Sci., 2005, 62, 2956–2973 CrossRef CAS PubMed.
- M. P. Chadwick, B. R. Westley and F. E. B. May, Biochem. J., 1997, 327, 117–123 CrossRef CAS PubMed.
- B. R. Westley, S. M. Griffin and F. E. B. May, Biochemistry, 2005, 44, 7967–7975 CrossRef CAS PubMed.
- T. Marchbank, B. R. Westley, F. E. B. May, D. P. Calnan and R. J. Playford, J. Pathol., 1998, 185, 153–158 CrossRef CAS PubMed.
- C. Bossenmeyer-Pourie, R. Kannan, S. Ribieras, C. Wendling, I. Stoll, L. Thim, C. Tomasetto and M. C. Rio, J. Cell Biol., 2002, 157, 761–770 CrossRef CAS PubMed.
- W. Hoffmann, Cell. Mol. Life Sci., 2005, 62, 2932–2938 CrossRef CAS PubMed.
- M. A. Williams, B. R. Westley, F. E. B. May and J. Feeney, FEBS Lett., 2001, 493, 70–74 CrossRef CAS PubMed.
- P. D. Rye, J. Keyte, M. J. Sutcliffe, K. Bailey and R. A. Walker, Protein Pept. Lett., 1994, 1, 54–59 CAS.
- E. C. B. Johnson and S. B. H. Kent, J. Am. Chem. Soc., 2006, 128, 6640–6646 CrossRef CAS PubMed.
- T. M. Hackeng, J. H. Griffin and P. E. Dawson, Proc. Natl. Acad. Sci. U. S. A., 1999, 96, 10068–10073 CrossRef CAS PubMed.
- M. Muttenthaler, F. Albericio and P. E. Dawson, Nat. Protoc., 2015, 10, 1067–1083 CrossRef CAS PubMed.
- J.-S. Zheng, S. Tang, Y.-K. Qi, Z.-P. Wang and L. Liu, Nat. Protoc., 2013, 8, 2483–2495 CrossRef CAS PubMed.
- H. B. Luo, M. Y. Cao, K. Newell, C. Afdahl, J. Wang, W. K. Wang and Y. L. Li, J. Chromatogr. A, 2015, 1424, 92–101 CrossRef CAS PubMed.
- F. C. Cardoso, Z. Dekan, J. J. Smith, J. R. Deuis, I. Vetter, V. Herzig, P. F. Alewood, G. F. King and R. J. Lewis, Br. J. Pharmacol., 2017, 174, 2528–2544 CrossRef CAS PubMed.
- J. S. Wingerd, C. A. Mozar, C. A. Ussing, S. S. Murali, Y. K. Chin, B. Cristofori-Armstrong, T. Durek, J. Gilchrist, C. W. Vaughan, F. Bosmans, D. J. Adams, R. J. Lewis, P. F. Alewood, M. Mobli, M. J. Christie and L. D. Rash, Sci. Rep., 2017, 7, 974–988 CrossRef PubMed.
- H. Takahashi, J. I. Kim, H. J. Min, K. Sato, K. J. Swartz and I. Shimada, J. Mol. Biol., 2000, 297, 771–780 CrossRef CAS PubMed.
- L. Thim, H. F. Woldike, P. F. Nielsen, M. Christensen, K. Lynchdevaney and D. K. Podolsky, Biochemistry, 1995, 34, 4757–4764 CrossRef CAS PubMed.
- M. D. Carr, Biochemistry, 1992, 31, 1998–2004 CrossRef CAS PubMed.
- D. S. Wishart, C. G. Bigam, A. Holm, R. S. Hodges and B. D. Sykes, J. Biomol. NMR, 1995, 5, 67–81 CrossRef CAS PubMed.
- M. Clyne, P. Dillon, S. Daly, R. O'Kennedy, F. E. May, B. R. Westley and B. Drumm, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 7409–7414 CrossRef CAS PubMed.
- E. P. Reeves, T. Ali, P. Leonard, S. Hearty, R. O'Kennedy, F. E. May, B. R. Westley, C. Josenhans, M. Rust, S. Suerbaum, A. Smith, B. Drumm and M. Clyne, Gastroenterology, 2008, 135, 2043–2054 CrossRef CAS PubMed.
- B. Dolan, J. Naughton, N. Tegtmeyer, F. E. B. May and M. Clyne, PLoS One, 2012, 7, e47300 CrossRef CAS PubMed.
- M. Clyne and F. E. B. May, Int. J. Mol. Sci., 2019, 20, 4400–4413 CrossRef CAS PubMed.
- R. Stürmer, S. Harder, H. Schlüter and W. Hoffmann, ChemBioChem, 2018, 19, 2598–2608 CrossRef PubMed.
- F. Heuer, R. Stürmer, J. Heuer, T. Kalinski, A. Lemke, F. Meyer and W. Hoffmann, Int. J. Mol. Sci., 2019, 20, 871–5883 CrossRef PubMed.
- R. Stürmer, J. Reising and W. Hoffmann, Int. J. Mol. Sci., 2019, 20, 6052–6060 CrossRef PubMed.
- F. G. Hanisch, H. Ragge, T. Kalinski, F. Meyer, H. Kalbacher and W. Hoffmann, Glycobiology, 2013, 23, 2–11 CrossRef CAS PubMed.
- L. Thim, F. Madsen and S. S. Poulsen, Eur. J. Clin. Invest., 2002, 32, 519–527 CrossRef CAS PubMed.
- C. K. Yu, C. J. Wang, Y. Chew, P. C. Wang, H. S. Yin and M. C. Kao, Biochem. Biophys. Res. Commun., 2016, 477, 794–800 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0cc02321c |
| This journal is © The Royal Society of Chemistry 2020 |