
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTriflyl-assisted reductive Pd-catalyzed Tsuji–Trost type reaction†
Carlos
Lázaro-Milla
 a,
M. Teresa
Quirós
b,
Diego J.
Cárdenas
b and
Pedro
Almendros
*c
a,
M. Teresa
Quirós
b,
Diego J.
Cárdenas
b and
Pedro
Almendros
*c
aGrupo de Lactamas y Heterociclos Bioactivos, Departamento de Química Orgánica, Unidad Asociada al CSIC, Facultad de Química, Universidad Complutense de Madrid, Madrid 28040, Spain
bDepartamento de Química Orgánica, Facultad de Ciencias, Institute for Advanced Research in Chemical Sciences, IAdChem, Universidad Autónoma de Madrid, Av. Francisco Tomás y Valiente 7, Cantoblanco, Madrid 28049, Spain
cInstituto de Química Orgánica General, IQOG-CSIC, Juan de la Cierva 3, Madrid 28006, Spain. E-mail: palmendros@iqog.csic.es
First published on 21st April 2020
Abstract
New (triflyl)cyclobutenes have been prepared by palladium-catalyzed hydrodetriflylation reaction using water and deuterium oxide as convenient hydrogen and deuterium sources. In addition, an investigation of the possible mechanism for this Tsuji–Trost type reaction of bis(triflyl)cyclobutenes has been facilitated by labelling studies and density functional theory (DFT) calculations.
The Tsuji–Trost reaction, which consists of a metal-catalyzed reaction of allylic substrates bearing a good leaving group (LG) with nucleophiles, is one of the most convenient methods for carbon–carbon and carbon–heteroatom bond formation (Scheme 1a).1 Carbon, nitrogen, oxygen, sulfur, phosphorous, and selenium nucleophiles successfully lead to the corresponding functionalized allylic products. Regarding allylic hydrogenolysis that could provide naked olefins, the most commonly used hydrogen transfer agents are typical reducing reagents such as aluminohydrides, borohydrides, hydrosilanes, tin hydrides, formic acid and ammonium formate (Scheme 1b).2 The utilization of these very reactive reagents leads to difficulties regarding selectivity and functional group compatibility. However, water is the cheapest and most environmentally friendly hydrogen source. Taking into account that recent reports pointed out the use of water as the hydrogen source in Pd-catalyzed hydrogenation reactions (Scheme 1c),3 we decided to study the feasibility of a novel palladium-catalyzed allylic substitution reaction using water as the hydrogen source as a direct route to (triflyl)cyclobutenes4 (Scheme 1d).
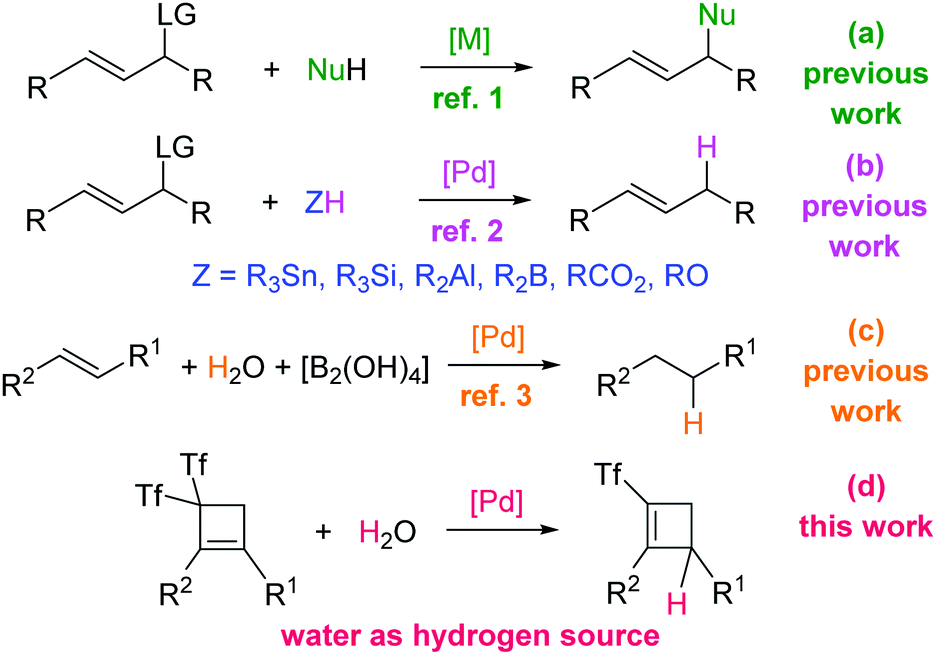 | ||
| Scheme 1 Mode of reaction between activated alkenes and nucleophiles: previous studies and the present work. | ||
The starting materials, cyclobutenes 1, were easily prepared from the corresponding alkynes through a formal [2+2] cyclization reaction using Yanai's reagent.5 Initial experiments were performed using cyclobutene 1a as a model substrate (Table 1). Pd(OAc)2, Pd(TFA)2, PdCl2, Pd(CH3CN)2Cl2, Pd(PPh3)2Cl2, Pd(amphos)Cl2, Pd(dba)2, Pd(dppf)2, and Pd(PPh3)4 were tested as the palladium salts. The above screening demonstrated that 5 mol% Pd(PPh3)4 was the best choice (Table 1, entries 1–9). A lower catalyst loading had a detrimental effect on the yield. Different bases (K2CO3, Cs2CO3, K3PO4, and Et3N) and solvents (THF, acetonitrile, DMF, and 1,4-dioxane) were tested, with the combination of K2CO3/1,4-dioxane providing the highest yield (93%; Table 1, entry 9). The integrity of the catalyst decreases under exposure to air, leading to diminished yields of (triflyl)cyclobutene 2a. The absence of (triflyl)cyclobutene 2a was noticed when either the Pd complex or water was not present.
|
|
|||
|---|---|---|---|
| Entry | Catalyst | Reaction conditions | Yielda (%) |
| a Yield of pure, isolated product with correct analytical and spectral data. | |||
| 1 | Pd(OAc)2 | K2CO3, 60 °C, 12 h | 45 |
| 2 | Pd(TFA)2 | K2CO3, 60 °C, 12 h | 48 |
| 3 | PdCl2 | K2CO3, 60 °C, 12 h | 23 |
| 4 | Pd(CH3CN)2Cl2 | K2CO3, 60 °C, 4 h | 73 |
| 5 | Pd(PPh3)2Cl2 | K2CO3, 60 °C, 5 h | 85 |
| 6 | Pd(amphos)Cl2 | K2CO3, 60 °C, 12 h | 40 |
| 7 | Pd(dba)2 | K2CO3, 60 °C, 3 h | 83 |
| 8 | Pd(dppf)2 | K2CO3, 60 °C, 12 h | — |
| 9 | Pd(PPh3)4 | K2CO3, 60 °C, 4 h | 93 |
| 10 | Pd(PPh3)4 | K2CO3, RT, 24 h | 5 |
| 11 | Pd(PPh3)4 | Et3N, 60 °C, 12 h | — |
| 12 | Pd(PPh3)4 | K3PO4, 60 °C, 6 h | 75 |
| 13 | Pd(PPh3)4 | Cs2CO3, 60 °C, 4 h | 81 |
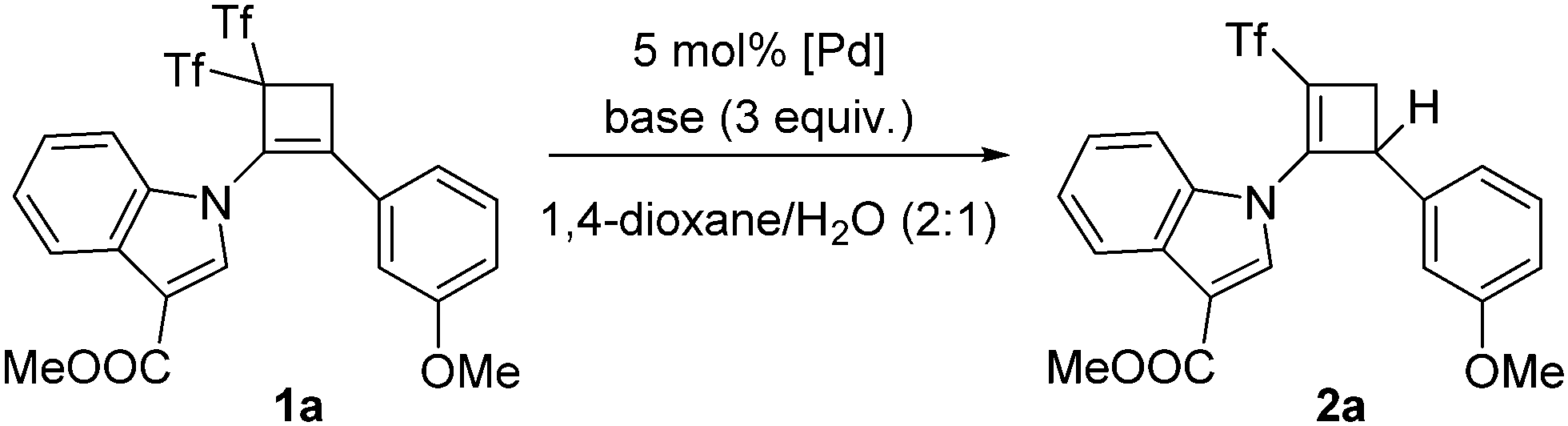
With the best conditions for obtaining adduct 2a in hand, the substrate scope was next studied. Thus, bis(triflyl)cyclobutenes 1a–i were smoothly converted into (triflyl)cyclobutenes 2a–i under the optimal reaction conditions (Scheme 2). The transformation tolerated aryl groups bearing substituents of different electronic properties such as methoxy (2a) and nitro (2b), as well as heteroaryl moieties such as indole (2a–d), carbazole (2e) and indazole (2f). Besides, heteroatomic substituents such as chlorine (2g), sulfone (2h), and phosphine oxide (2i) were well accommodated. As shown in Scheme 2, the allylic substitution reaction is totally regioselective because isomeric products 2-isomer-a–i were not detected. However, bis(triflyl)cyclobutenes 1j–l bearing a lactam substituent reacted to give unexpected cyclobutenones 3j–l (see ESI† for a plausible reaction path for the formation of products 3) rather than (triflyl)cyclobutenes 2j–l (Scheme 2). It is worth noting that the palladium-catalyzed hydrodetriflylation reaction of bis(triflyl)cyclobutenes 1m–s bearing an alkyl substituent resulted in the formation of (triflyl)cyclobutenes 4m–s having an extra alkene functionality (Scheme 3). Olefins 4m–s were obtained with total stereoselectivity as single E-isomers. At first glance, the formation of the exocyclic alkene moiety in 1,3-dienes 4m–s can be viewed as a formal molecular hydrogen release6 in non-detected (triflyl)cyclobutenes 2m–s. Of particular interest was the reactions of bis(triflyl)cyclobutenes 1t–v, which were converted into 1,4-dienes 5t,u and cyclobutenal 5v by the evolution of 1,3-dienes 4t–vvia a 1,3-H shift (Scheme 4). This attractive synthesis sequence constitutes a striking example of the versatility of the above palladium-catalyzed reaction in organic synthesis.
 | ||
| Scheme 2 Palladium-catalyzed preparation of (triflyl)cyclobutenes 2 using water as the hydrogen source. | ||
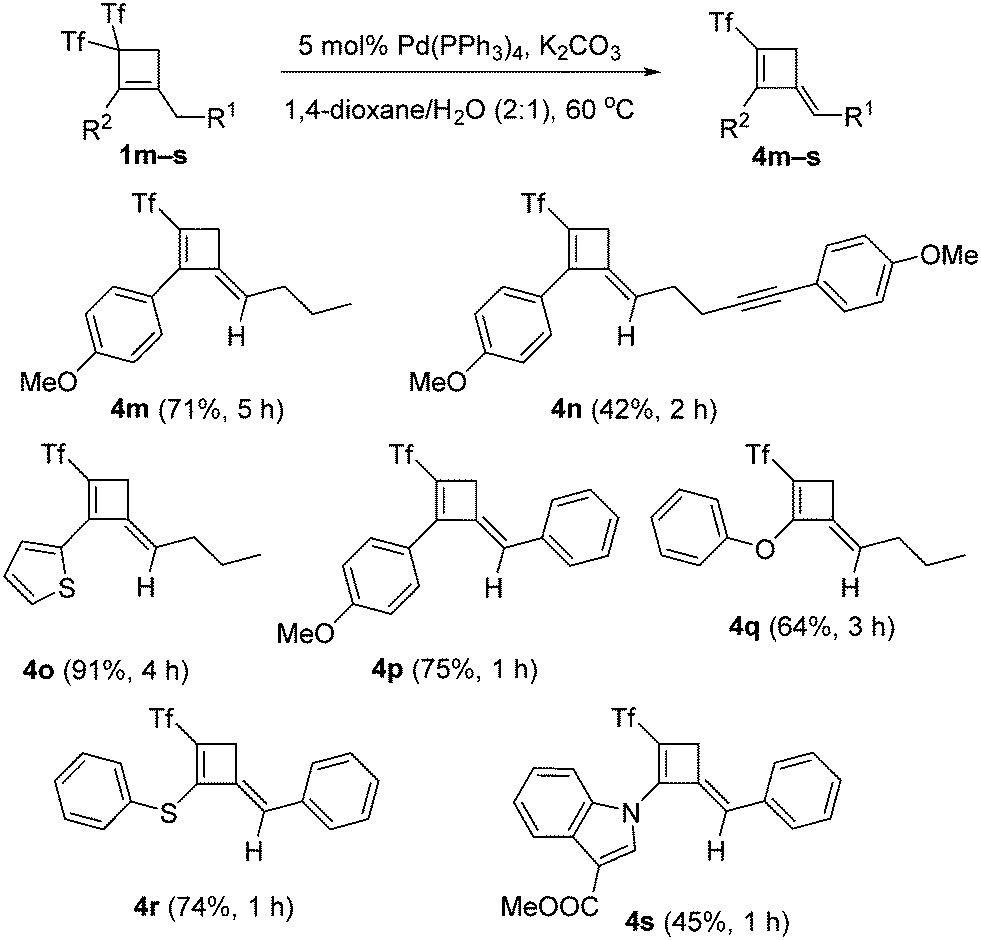 | ||
| Scheme 3 Palladium-catalyzed preparation of (triflyl)cyclobutenes 4. | ||
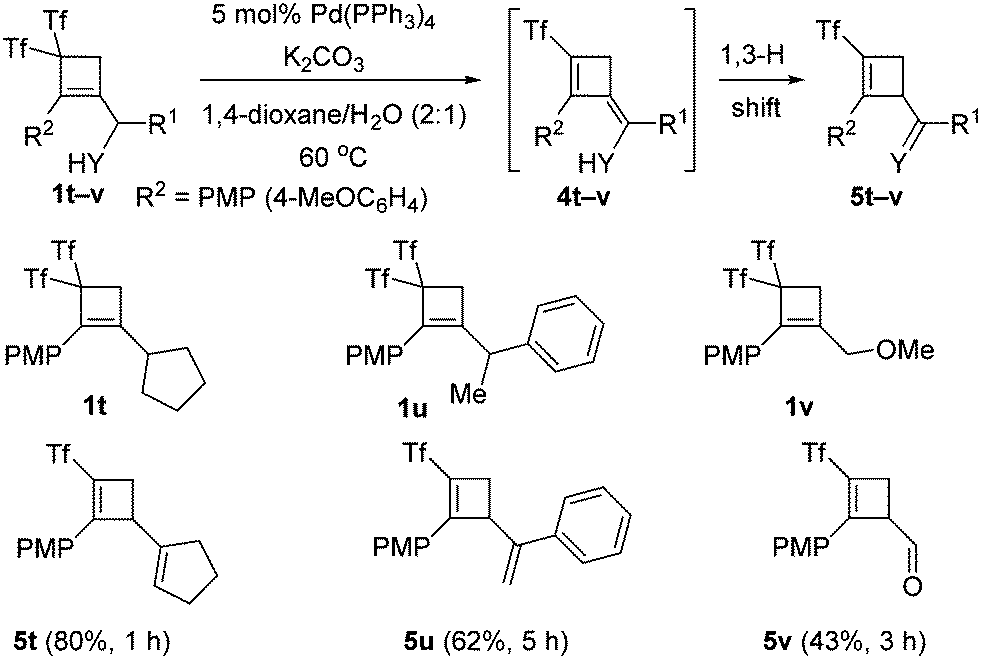 | ||
| Scheme 4 Palladium-catalyzed preparation of (triflyl)cyclobutenes 5. | ||
To gain a better understanding of the hydrodetriflylation process, control and deuterium-labelling experiments were carried out. Under otherwise identical conditions, adduct 2a was obtained in a similar yield independent of the presence or absence of TEMPO as an additive (Scheme 5), which probably ruled out a radical mechanism.7 When bis(triflyl)cyclobutene 1a was treated under the optimal conditions using D2O instead of H2O, (triflyl)cyclobutene [D]-2a was obtained with 91% deuterium incorporation (Scheme 5).8 Thus, the deuterium atom can be easily incorporated into the cyclobutene ring by the current hydrodetriflylation reaction, which uses inexpensive water as the stoichiometric hydrogen source. Bis(triflyl)cyclobutene [D2]-1a with total deuterium incorporation at the methylenic carbon of the small ring was prepared from the novel deuterated Yanai's reagent [D2]-Y (see ESI† for details). It was observed that the reactions of bis(triflyl)cyclobutene [D2]-1a with H2O and D2O formed deuterium-labeled (triflyl)cyclobutenes [D2]-2a and [D3]-2a, respectively (Scheme 5). As expected, no deuterated cyclobutene 4p was detected starting from 1p (Scheme 5).
 | ||
| Scheme 5 Control and deuterium labelling experiments. | ||
We performed a detailed computational study at the DFT level on the possible catalytic reaction pathways that may account for the formation of cyclobutene-triflones 2 and 4 from bis(triflyl)cyclobutenes 1 in the presence of water (Scheme 6).9 The bis(triflyl)cyclobutene with the alkene substituted with Ph and Et groups was used as the model substrate, and (PPh3)Pd was used as the catalytically active species.10 All the computed energies are compatible with a process that takes place at 60 °C. The first step of the catalytic cycle is the coordination of palladium to the alkene moiety of bis(triflyl)cyclobutenes 1. This reaction is a favorable exergonic process (ΔG = −18.8 kcal mol−1) and gives the intermediate 1-Pd. The oxidative addition to form π-allyl Pd(II) I is endergonic (ΔG = 5.6 kcal mol−1) and occurs with a moderate activation energy of 15.4 kcal mol−1. Therefore, the two step process leading to the allyl-Pd intermediates proceeds downhill. An alternative stepwise reaction in which π-allyl Pd(II) I is formed through oxidative addition paired with the liberation of Tf− that then coordinates with Pd is highly unfavorable (see ESI† for details). The formation of two different σ-allyl Pd(II) complexes, II and III, is feasible from I, with the energy difference of 1.8 kcal mol−1. Interestingly, II is more stable in the form that presents the triflyl group coordinated through one of the oxygen atoms, whereas the form of III that has the lower energy is the one in which the triflyl group is coordinated through the sulfur atom (see ESI† for details). The difference in energy between the coordination bond isomers is ∼5 kcal mol−1, pointing to an equilibrium in solution between these species. The fact that both σ-allyl Pd(II) complexes are higher in energy than I is possibly due to the generation of a coordinative vacancy in Pd. Compared to the usual stabilization of the corresponding σ-allyl Pd(II) complexes upon coordination with phosphine, in this case, the addition of an additional PPh3 to the coordination sphere of palladium increases the energy of these σ-allyl Pd(II) complexes. However, in the reaction medium, the vacant coordination site formed after Pd slipping to the η1-coordination mode could be occupied. In fact, the coordination of water stabilizes the corresponding σ-allyl Pd(II) complexes (see ESI† for details).
 | ||
| Scheme 6 Plausible pathways for the formation of (triflyl)cyclobutenes 2 (Cycle A) and 4 (Cycle B). The values of reaction free energies and activation energies (kcal mol−1) were calculated at the B3LYP/6-31G(d) (C,H,O,S,F,P,K) and LANL2DZ (Pd) level in the gas phase with R1 = Me, R2 = Ph, and [Pd] = (PPh3)Pd. | ||
Noticeably, calculations show that starting from each σ-allyl Pd(II) complex, a different reaction pathway is preferred (Scheme 7). From II, protodemetallation with water to give IV is an almost thermoneutral process (ΔG = 3.8 kcal mol−1) viaTSII–IV (ΔG‡ = 17.7 kcal mol−1). In this step, a proton from water bonds to the carbon of the alkene that is further from Pd, forming at the same time a Pd-hydroxide, which remains coordinated to the newly formed double bond. The remaining steps to end the catalytic cycle are both exergonic (ΔG = −13.5 and −11.9 kcal mol−1, respectively). These steps imply the formation of Pd(0) species by a formal reductive elimination of TfOH, which may take place by external water attack on coordinated Tf rather than the direct formation of a S–O bond from IV.
 | ||
| Scheme 7 Comparison of both possible reaction pathways from π-allyl Pd(II) complex I to form σ-allyl Pd(II) complex II (path A) or III (path B). Reaction free energies and activation energies (kcal mol−1) are calculated at the B3LYP/6-31G(d) (C,H,O,S,F,P,K) and LANL2DZ (Pd) level in the gas phase. | ||
In contrast to the evolution of II, we were not able to locate a low energy TS for the direct cleavage of the Pd–C bond in III with water. However, the β-hydride elimination to form product 4 and palladium hydride V was found to be an exergonic process (ΔG = −4.7 kcal mol−1) with a moderately low activation energy (ΔG‡ = 12.7 kcal mol−1) (path B, Scheme 7). It is important to note that stabilization of the transition states for both protodemetallation and β-hydrogen elimination is achieved by water coordination along the reaction pathway and, therefore, the calculated activation energies are upper limits and the actual barriers are probably lower. Furthermore, an overall calculation of the reductive elimination step assisted by K2CO3 from V to regenerate the Pd(0) catalytically active species shows that the process is, as expected, highly exergonic (ΔG = −49.7 kcal mol−1) (Scheme 6).11,12 According to these results, path B would be the preferred reaction pathway, since the energy barrier is lower than the alternative path A. Path A would occur in bis(triflyl)cyclobutenes 1 in which the β-hydride elimination process is not attainable.
In summary, a palladium-catalyzed synthesis of (triflyl)cyclobutenes has been developed through an allylic substitution reaction, taking advantage of the use of water as the hydrogen source. This method can also be used for the preparation of deuterated cyclobutenes with inexpensive D2O as the deuterium source. Labeling experiments and DFT calculations support the proposal of a possible pathway for this Pd-catalyzed water assisted hydrogen transfer reaction.
This work was supported in part by AEI (MICIU) and FEDER (Projects PGC2018-095025-B-I00 and CTQ2016-79826-R). M. T. Q. thanks MICIU for a Juan de la Cierva contract. C. L. M. thanks MICIU and UCM for a postdoctoral contract. We thank the Centro de Computación Científica-UAM for computational time. We are grateful to Prof. B. Alcaide for his continued support.
Conflicts of interest
There are no conflicts of interest to declare.Notes and references
- For selected reviews, see: (a) B. M. Trost and C. A. Kalnmals, Chem. – Eur. J., 2020, 26, 1906 CAS; (b) S. Parisotto and A. Deagostino, Synthesis, 2019, 1892 CAS; (c) Y.-N. Wang, L.-Q. Lu and W.-J. Xiao, Chem. – Asian J., 2018, 13, 2174 CrossRef CAS PubMed; (d) R. L. Grange, E. A. Clizbe and P. A. Evans, Synthesis, 2016, 2911 CAS; (e) B. M. Trost and M. L. Crawley, Chem. Rev., 2003, 103, 2921 CrossRef CAS PubMed.
- For selected reviews, see: (a) A. Heumann, in Transition Metal for Organic Synthesis, ed. M. Beller and C. Bolm, Wiley-VCH, Weinheim, 2004, pp. 307–320 Search PubMed; (b) J. Tsuji and T. Mandai, Synthesis, 1996, 1 CrossRef CAS.
- (a) S. P. Cummings, T.-N. Le, G. E. Fernandez, L. G. Quiambao and B. J. Stokes, J. Am. Chem. Soc., 2016, 138, 6107 CrossRef CAS PubMed; (b) Q. Xuan and Q. Song, Org. Lett., 2016, 18, 4250 CrossRef CAS; (c) W. Kong, Q. Wang and J. Zhu, Angew. Chem., Int. Ed., 2017, 56, 3987 CrossRef CAS PubMed; (d) S. Rao and K. R. Prabhu, Chem. – Eur. J., 2018, 24, 13954 CrossRef CAS PubMed; (e) J. Cui, L. Meng, X. Chi, Q. Liu, P. Zhao, D.-p. Zhang, L. Chen, X. Li, Y. Donga and H. Liu, Chem. Commun., 2019, 55, 4355 RSC; (f) A. G. Campaña, R. E. Estévez, N. Fuentes, R. Robles, J. M. Cuerva, E. Buñuel, D. J. Cárdenas and J. E. Oltra, Org. Lett., 2007, 9, 2195 CrossRef PubMed.
- The cyclobutene core is the key motif of several bioactive compounds and advanced materials. (a) S. Ohno, R. F. Avena, H. Aoyama, H. Fujioka and M. Arisawa, Green Chem., 2020, 22, 1220 RSC; (b) C. Zhong, Y. Huang, H. Zhang, Q. Zhou, Y. Liu and P. Lu, Angew. Chem., Int. Ed., 2020, 59, 2750 CrossRef CAS PubMed; (c) S. Feng, H. Hao, P. Liu and S. L. Buchwald, ACS Catal., 2020, 10, 282 CrossRef CAS; (d) B. Jiang, Y. Wei and M. Shi, Org. Chem. Front., 2018, 5, 2091 RSC; (e) M. Eisold, A. N. Baumann, G. M. Kiefl, S. T. Emmerling and D. Didier, Chem. – Eur. J., 2017, 23, 1634 CrossRef CAS PubMed.
- B. Alcaide, P. Almendros, I. Fernández and C. Lázaro-Milla, Chem. Commun., 2015, 51, 3395 RSC.
- For α,β-dehydrogenation of carbonyls via palladium catalysis, see: Y. Zhao, Y. Chen and T. R. Newhouse, Angew. Chem., Int. Ed., 2017, 56, 13122 CrossRef CAS.
- For palladium-catalyzed hydrogen-transfer radical cyclization, see: M. Ratushnyy, M. Parasram, Y. Wang and V. Gevorgyan, Angew. Chem., Int. Ed., 2018, 57, 2712 CrossRef CAS PubMed.
- Deuterated organic compounds are valuable in the search for new drugs due to the improvement of the pharmacological properties. For reviews, see: (a) J. Atzrodt, V. Derdau, W. J. Kerr and M. Reid, Angew. Chem., Int. Ed., 2018, 57, 1758 CrossRef CAS PubMed; (b) T. Pirali, M. Serafini, S. Cargnin and A. A. Genazzani, J. Med. Chem., 2019, 62, 5276 CrossRef CAS PubMed.
- Initially, a mechanism involving the assistance of the triflyl group in the use of water as a hydrogen source was envisaged. In this manifold, water would attack to the coordinated sulphur atom in III, followed by a proton shift to form a Pd–H intermediate. However, the calculated energy for the intermediates of this alternative mechanism was high above III (ΔG = 35.8 kcal mol−1), which would be incompatible with a productive reaction outcome (see ESI† for details).
- (a) M. Ahlquist, P. Fristrup, D. Tanner and P.-O. Norrby, Organometallics, 2006, 25, 2066 CrossRef CAS; (b) K. Wang, Y. Lu, F. Hu, J. Yang, Y. Zhang, Z.-X. Wang and J. Wang, Organometallics, 2018, 37, 1 CrossRef CAS.
- I. D. Hills and G. C. Fu, J. Am. Chem. Soc., 2004, 126, 13178 CrossRef CAS PubMed.
- For a detailed study on the reductive elimination assisted by K2CO3 on Pd–H see: D. B. Eremin, E. A. Denisova, A. Y. Kostyukovich, J. Martens, G. Berden, J. Oomens, V. N. Khrustalev, V. M. Chernyshev and V. P. Ananikov, Chem. – Eur. J., 2019, 25, 16564 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Computational details, experimental procedures, characterization data of new compounds, and copies of NMR spectra for all new compounds. See DOI: 10.1039/d0cc02146f |
| This journal is © The Royal Society of Chemistry 2020 |