
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Unveiling the reaction mechanism of novel copper N-alkylated tetra-azacyclophanes with outstanding superoxide dismutase activity†
Álvaro
Martínez-Camarena
 *a,
Pedro A.
Sánchez-Murcia
*b,
Salvador
Blasco
b,
Leticia
González
bc and
Enrique
García-España
a
*a,
Pedro A.
Sánchez-Murcia
*b,
Salvador
Blasco
b,
Leticia
González
bc and
Enrique
García-España
a
aICMol, Departamento de Química Inorgánica, University of Valencia, C/Catedrático José Beltrán 2, 46980, Paterna, Spain. E-mail: alvaro.martinez@uv.es
bInstitute of Theoretical Chemistry, Faculty of Chemistry, University of Vienna, Währinger Str. 17, A-1090 Vienna, Austria. E-mail: pedro.murcia@univie.ac.at
cVienna Research Platform for Accelerating Photoreaction Discovery, University of Vienna, Währinger Str. 17, 1090 Vienna, Austria
First published on 28th May 2020
Abstract
Quantum chemical and multiscale calculations reveal the mechanistic pathway of two superoxide dismutase mimetic N-alkylated tetra-azacyclophane copper complexes with remarkable activity. The arrangement of the binding site afforded by the bulky alkyl substituents and the coordinated water molecule as a proton source play key roles in the reaction mechanism.
The use of O2 in the metabolism of aerobic organisms is associated with the generation of toxic reactive oxygen species (ROS).1 The imbalance between their production and clearance leads to oxidative stress, thereby associated with the possible development of diseases.2 Since the superoxide anionic radical (O2−) is the first ROS species formed in the O2 reductive pathway, the removal of its metabolic excesses is crucial to avoid oxidative stress. To do so, living organisms are equipped with superoxide dismutase enzymes (SODs), which transform O2− into molecular oxygen (O2) and hydrogen peroxide (H2O2).3 Inspired by nature, many efforts have been carried out to mimic the SOD activity with small molecules.4 Previous studies showed that polyazacyclophane receptors are capable of binding free metal ions, preventing ROS production and having protective capacity against ROS.1,4c,5 However, to the best of our knowledge, no compound has been approved yet for therapeutic intervention.4b
With the aim to build up improved human SOD (hSOD) mimetics, we recently reported6 the design and synthesis of complex [CuL1(OH2)]2+1 (Fig. 1A and B). In 1, Cu2+ is complexed in the equatorial plane by the two nitrogen atoms of L1 (Fig. 1B) alkylated with isopropyl groups, by the pyridine nitrogen and by a water molecule, while the axial position is occupied by the methyl-alkylated nitrogen placed at the middle of the polyamine bridge of L1.7 Complex 1 shares with the active site of Cu in human SOD1 and SOD3 (hSOD1/hSOD3, in Fig. 1C) the fact that its coordination sphere is composed by four N-donors and a water molecule. Moreover, the crystallographic structure of 1 matches very nicely the square-pyramidal geometry of the electroactive site of the enzyme (Fig. 1D).
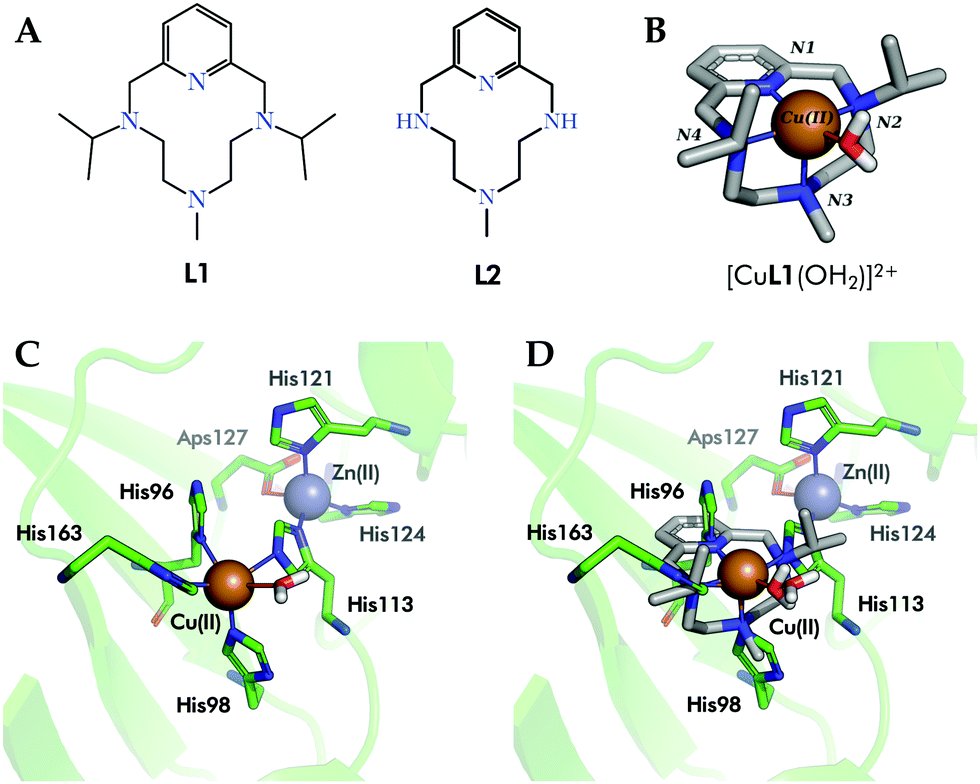 | ||
| Fig. 1 (A) Ligands L1 and L2 are the macrocycle in complexes 1 and 2, respectively. (B) Crystal structure of complex [CuL1(OH2)]2+ (1), CCDC id. 1827336. (C) Detail of the active center of hSOD1 (PDB id. 2JLP). (D) Superimposition of complex 1 (sticks, carbon atoms colored in grey) within the active site of hSOD1 (green, PDB id. 2JLP). Only the inner sphere residues have been shown for simplicity. | ||
Here we report the in vitro SOD activity of 1 and unveil one of the first ever reported all-atom catalytic mechanisms for SOD mimetics. Using quantum mechanics (QM) and hybrid quantum chemistry molecular mechanics (QM/MM) methods we show that, similar to the native SOD enzymes containing either Cu, Mn or Fe as electroactive metals, the coordinated water molecule plays a key role in the catalytic mechanism as a proton source and sink.
The in vitro SOD activity of 1 was measured at the physiological pH of 7.4 by means of the enzymatic assay developed by McCord–Fridovich.8 We also designed and tested complex [CuL2(OH2)]2+ (2) (see Fig. S10–S12, Table S1 and Table 1 with the potentiometric data in the ESI†), where the isopropyl substituents are absent (Fig. 1A). Although the bulkiness of the isopropyl substituents produces a slight decrease in the stability of the complexes,6 both ligands form very stable Cu2+ complexes at neutral pH. The SOD activities (kcat values of 13.7 × 106 M−1 s−1 for 1 and 7.2 × 106 M−1 s−1 for 2) are among the highest reported so far for synthetic SOD mimetics.9 Remarkably, the activities of 1 and 2 are only one order of magnitude below that of native hSOD1 (43.0 × 107 M−1 s−1).6,10 It is interesting to remark that the best performance is observed for the complex of the ligand with bulkier groups.
Encouraged by these results, we considered it of relevance to model the reaction mechanism of disproportionation of superoxide in the presence of copper complexes in order to identify the molecular features that govern the catalytic activity. Previous experimental kinetic studies showed that the disproportionation reaction is based on two elemental reactions:11
| L–Cu(II) + O2˙− → L–Cu(I) + O2 | (1) |
| L–Cu(I) + O2˙− + 2H+ → L–Cu(II) + H2O2 | (2) |
Based on initial explorations, we assumed an associative mechanism as an initial hypothesis for both half-reactions (Fig. 2A), and we chose B3LYP-D3/def2-svp/Cu(MDF10) as level of theory (see Section IV in the ESI†) due to the good correlation between the obtained structures and the experimental ones. The solvent was implicitly treated with the polarizable continuum model. Fig. 2B shows the computed structures and energies of the catalytic cycle for 1. Similar to SOD,14 it consists of two half-reactions (see also Fig. 2A): in the first one, Cu2+ is reduced to Cu+ by oxidation of superoxide to molecular oxygen; in the second one, a new superoxide anion oxidizes Cu+ to Cu2+ requiring two protons and releasing a molecule of hydrogen peroxide. In turn, each half-reaction may consist of three main steps: (i) the approach of the superoxide radical to the coordination sphere (π-complex 1-O2˙−, intermediate B), (ii) the binding of O2˙− within the copper coordination sphere (intermediates A or D), and finally, (iii) the leaving of products of the reaction (O2 in complexes A or the peroxide in complexes D).
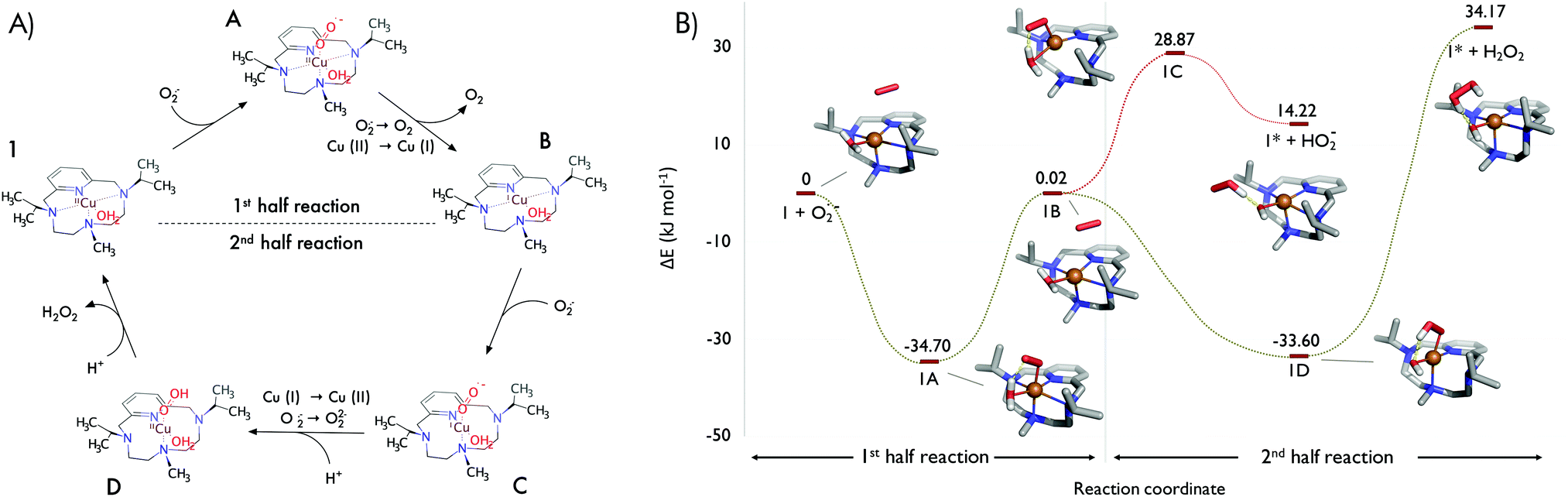 | ||
| Fig. 2 (A) Proposed associative reaction mechanism for the catalysed disproportion of superoxide by 1: the oxidation of superoxide to O2 (1st half reaction) and the reduction of a second molecule of superoxide to H2O2 (2nd half reaction). (B) Energy profile (kJ mol−1, B3LYP-D3/def2-svp/Cu(MDF10), calculated in water using the SCRF method of the polarizable continuum model) for the reaction mechanism of the catalytic oxidation and reduction of two species of superoxide by complex 1. Structure 1* corresponds to the hydroxylated form of 1. | ||
In agreement with SOD enzymes,11 our calculations predict that the first half-reaction is barrierless. No transition state was identified in the whole path (Fig. 2B). We also observed that the oxidation state of the metal centre strongly affects the geometry of the coordination sphere. In the first part of the reaction, the superoxide enters by forming a metastable π-complex that evolves into the intermediate 1A in a process that releases energy (34.70 kJ mol−1). The expansion of the coordination number of Cu2+ from 5 to 6 in 1A corroborates that the first half reaction seems to follow an associative mechanism. Hexacoordinated intermediate 1A has a distorted octahedral molecular geometry with the N2 and N4 (recall labelling in Fig. 1B) atoms in the axial positions. Remarkably, the superoxide enters into the coordination sphere by establishment of a hydrogen bond with the coordinated water molecule, which stabilizes 1A in energy over 10 kJ mol−1 (data not shown).
In the second half-reaction, the entering superoxide is reduced to hydrogen peroxide (Fig. 2A). Therefore, here the two protons should be provided by water molecules as no other ionizable groups are available in the surroundings. In order to select extra water molecules, we run QM/MM-MD simulations with complex 1B (oxidation state Cu+ in the presence of a superoxide molecule and embedded in a box of water molecules, see Materials and methods). After a few picoseconds of simulation, the superoxide coordinates the metal centre from the face defined by the pyridine ring and opposite to the coordinated water molecule to form the associative intermediate 1C (Fig. 2B). In such a disposition, the other oxygen of the superoxide is hydrogen-bonded to the coordinated water molecule and thus the first protonation is mediated by the coordinated water molecule (1D). The energy gap between 1C and 1D suggests that the entrance of the superoxide may be assisted by the proton transfer step. In addition, since the release of the monoprotonated peroxide is favourable (complex 1* + HO2−), the second protonation may also occur with the peroxide within the coordination sphere. Thus, the first protonation may follow an associative mechanism, with the coordinated water molecule as the general acid species to form the hydroxo intermediate 1D. The finding of a hydroxo intermediate is supported by the crystal structure of E. coli Mn-SOD (PDB id. 1IX9 and 1IXB) in which the Mn3+ is postulated to be bound to a hydroxo ligand.15 Previous QM computational studies of Fe-13a and Mn-SODs13 support the idea that the relevant forms of Fe3+-SOD and Mn3+-SOD are the hydroxo intermediates.7
The second protonation step to generate the hydrogen peroxide from 1D must involve a second water molecule from the solvent, in contrast to hSOD1, where the leaving of the peroxide is preceded by its protonation by a His residue to yield H2O2.12,16 It must be stressed that the second protonation is a highly endothermic step in our calculations (>60 kJ mol−1). Since the catalyst requires a third protonation to be regenerated into complex 1, this final protonation may occur before or together with the release of H2O2. Therefore, the coordinated water molecule (or its hydroxo counterpart) plays a pivotal role in the entering and leaving of the substrate.
In order to prove that the coordinated water molecule does not leave the coordination sphere and acts as a general acid, 1 was reacted with NaN3 to replace the coordinated water molecule by N3− before running the catalytic reaction. The determination of the interaction constant of N3− with [CuL1(OH2)]2+ confirmed its capacity to occupy the fifth coordination position in the complex (log K = 3.05(1)), substituting the coordinated water as evidenced by the crystal structure in Fig. 3 and Fig. S13–S16 (ESI†). The measurements showed that the addition of NaN3 to a solution of [CuL1(OH2)]2+ leads to a 4-fold reduction of its catalytic constant from 13.7 × 106 to 3.7 × 106 M−1 s−1 (when 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1:5 Cu2+:L:N3− molar ratio is used). These results support our hypothesis that the water molecule (or its hydroxo form) remains bound to Cu throughout the whole catalytic cycle and acts as a general acid. With N3− in the coordination sphere, there is no water bound to the metal centre, the proton has to be acquired from the first water shell and thereby the catalytic activity drops.
:1:5 Cu2+:L:N3− molar ratio is used). These results support our hypothesis that the water molecule (or its hydroxo form) remains bound to Cu throughout the whole catalytic cycle and acts as a general acid. With N3− in the coordination sphere, there is no water bound to the metal centre, the proton has to be acquired from the first water shell and thereby the catalytic activity drops.
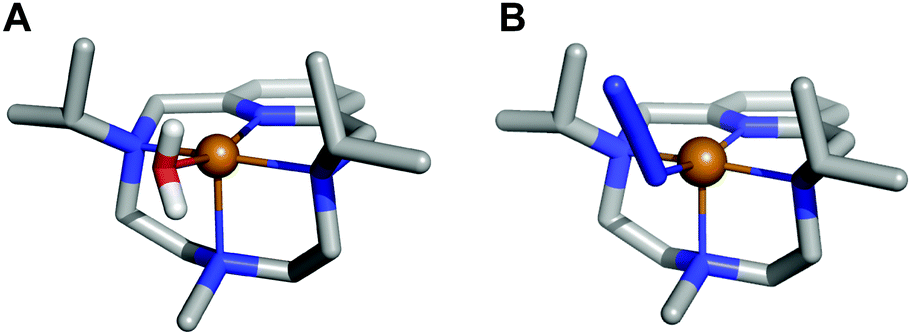 | ||
| Fig. 3 (A) Crystal structure of complex [CuL1(OH2)]2+ (1), CCDC id. 1827336. (B) Crystal structure of complex [CuL1(N3)]+, CCDC id. 1982975. | ||
For the second half reaction, if we compare complexes 1C (path not favourable) and 1D, there are significant differences between their structures. In 1C, L1 coordinates the copper atom just through N1 and N4, while in 1D the L1 the chelating atoms are N1 and N3 (atoms N2 and N4 do not coordinate now the copper centre). Intermediate 1C shows a highly distorted tetrahedral geometry (τ4 = 0.61), whereas intermediate 1D presents a smaller τ4 value (0.12), which can be related to a distorted square planar geometry (τ4 = 0 square planar; τ4 = 1 tetrahedral geometry).17 Thus, associated with the reduction of the superoxide radical in 1D, there is a conformational change in the adduct that leads to a square planar geometry from a square-pyramid one (1B).
We also computed the energy profile of complex 2, where the two isopropyl groups on nitrogens N2 and N4 were removed (Fig. S17, ESI†). 2 follows a similar trend as 1. However, the intermediate 2A in the first-half reaction is much more stabilized in energy than 1A. Since this process is barrier-less and this step shows the lowest energy of the reaction, the kinetics should be governed by the relative energy of the former two intermediates. This can be rationalized by an over stabilization of the catalyst:substrate adduct in 2 by the formation of hydrogen bonds not only with the water molecule complexed to the copper, but also with the hydrogen atoms belonging to the secondary amines of L2 (see Fig. S18 in the ESI†). This explains why the turnover of the catalyst decreases,18 as escaping from this minimum costs more energy than in complex 1A. This hypothesis is confirmed by the experimental measurements of the catalytic constant of 2, which is half of the value of 1. The second-half reaction in 2 is like that of 1.
Finally, we also analysed the dynamical behaviour of both complexes in aqueous solution by means of four QM/MM MD simulations considering both oxidation states (Cu2+ and Cu+) in the presence or absence of a superoxide molecule (see the ESI†). In the absence of superoxide, the alkylation of N2 and N4 with isopropyl groups in 1 reduces the Cu–N2/N3/N4 bond order regardless of the oxidation state (Table S2, ESI†). This means that the Cu–N bonds are longer in 1 than that in 2, with a broader distribution of the N–Cu bond distances along the simulations (the mean value for the Cu–N4 bond in 1 is 0.2 Å longer than that in 2, with a broader distribution along the simulation). To a minor extent, the Cu–N2 and Cu–N3 distances also increase in 1. Fig. 4 shows the superimposition of the structures obtained for each one of the complexes along the QM/MM MD trajectory. Furthermore, the evolution of the Cu–N2 distance for the complexes in their different oxidation states along the catalytic reaction mechanism is shown in Fig. S19 (ESI†). Clearly, in 2 the Cu–N2 bond order increases along all the catalytic cycle. The same feature is observed for the Cu–N3 and Cu–N4 bonds (Tables S6–S9, ESI†). These observations agree with the stability constants determined by the potentiometric titrations (Table 1). The reduction of four orders of magnitude of the Cu2+ complexation constant for L1 with respect to L2 is in agreement with the fact that its coordinative centre shows a broader distribution of bond distances and a higher average value. Thus, isopropylation of N2 and N4 in complex 1 increases the weakness of the N–Cu bonds and helps the complex to switch easily between the geometries implicated in the catalytic cycle, increasing the catalytic constant. The steric factors introduced by the isopropyl groups may also control the entrance of the superoxide to the coordination sphere by one face of the complex (Fig. S20, ESI†).
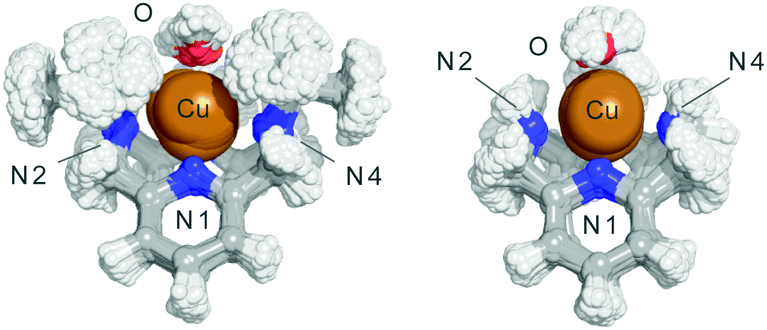 | ||
| Fig. 4 Superimposition of structures of complexes 1 and 2 obtained along the QM/MM MD simulations. | ||
In summary, this work reports the in vitro SOD catalytic activities of two N-alkylated tetra-azacyclophane copper complexes with remarkable antioxidant activities. Their mode of action, transforming the two superoxide molecules into oxygen and hydrogen peroxide in a catalysed cycle is put forward for the first time. It is observed that the isopropylation of the nitrogen atoms of the azacyclophane ring facilitates the structural changes required along the catalytic cycle and helps to orientate the entrance of the reactive species into the coordination sphere. Moreover, we confirm, both theoretically and experimentally, the key role of the coordinated water in the catalytic cycle of such SOD mimetics.
A. M.-C. thanks the Spanish Ministry of Science, Innovation and Universities for grants FPU14/05098 and EST16/00533. P. A. S.-M. and L. G. thank the University of Vienna, the Austrian FWF for financial support (Lise Meitner Project M 2260), and the Vienna Scientific Cluster (VSC3) for computational time. MINECO is acknowledged for the grants Juan de la Cierva (SB) and CTQ2016-78499-C6-1-R, CTQ2017-90852-REDC and Unidad de Excelencia María de Maeztu MDM-15-0538.
Conflicts of interest
There are no conflicts to declare.Notes and references
- K. M. Lincoln, P. Gonzalez, T. E. Richardson, D. A. Julovich, R. Saunders, J. W. Simpkins and K. N. Green, Chem. Commun., 2013, 49, 2712 RSC.
- L. R. Perez and K. J. Franz, Dalton Trans., 2010, 39, 2177 RSC.
- Y. Sheng, I. A. Abreu, D. E. Cabelli, M. J. Maroney, A. F. Miller, M. Teixeira and J. S. Valentine, Chem. Rev., 2014, 114, 3854 CrossRef CAS PubMed.
- (a) S. Gandhi and A. Y. Abramov, Oxid. Med. Cell. Longevity, 2012, 2012, 11 Search PubMed; (b) L. E. Scott and C. Orvig, Chem. Rev., 2009, 109, 4885 CrossRef CAS PubMed; (c) A. Martínez-Camarena, E. Delgado-Pinar, C. Soriano, J. Alarcón, J. M. Llinares, R. Tejero and E. Garcıa-Espana, Chem. Commun., 2018, 54, 3871 RSC; (d) D. P. Riley, Chem. Rev., 1999, 99, 2573 CrossRef CAS PubMed; (e) D. Salvemini, D. P. Riley and S. Cuzzocrea, Nat. Rev. Drug Discovery, 2002, 1, 367 CrossRef CAS PubMed.
- D. E. Green, M. L. Bowen, L. E. Scott, T. Storr, M. Merkel, K. Böhmerle, K. H. Thompson, B. O. Patrick, H. J. Schugar and C. Orvig, Dalton Trans., 2010, 39, 1604 RSC.
- Á. Martínez-Camarena, A. Liberato, E. Delgado-Pinar, A. G. Algarra, J. Pitarch-Jarque, J. M. Llinares, M. Á. Mañez, A. Domenech-Carbó, M. G. Basallote and E. García-España, Inorg. Chem., 2018, 57, 10961 CrossRef PubMed.
- J. Azadmanesh and G. E. O. Borgstahl, Antioxidants, 2018, 7, 25 CrossRef PubMed.
- (a) C. Beauchamp and I. Fridovich, Anal. Biochem., 1971, 44, 276 CrossRef CAS PubMed; (b) J. Y. Zhou and P. Prognon, J. Pharm. Biomed. Anal., 2006, 40, 1143 CrossRef CAS PubMed.
- (a) O. Iranzo, Bioorg. Chem., 2011, 39, 73 CrossRef CAS PubMed; (b) I. Batinić-Haberle, J. S. Rebouças and I. Spasojević, Redox-Active Therapeutics, Springer, 2016 CrossRef.
- H. Ohtsu, Y. Shimazaki, A. Odani, O. Yamauchi, W. Mori, S. Itoh and S. Fukuzumi, J. Am. Chem. Soc., 2000, 122, 5733 CrossRef CAS.
- (a) J. Rabani, D. Klug and I. Fridovich, Isr. J. Chem., 1972, 10, 1095 CrossRef CAS; (b) D. Klug-Roth, J. Rabani and I. Fridovich, J. Am. Chem. Soc., 1973, 95, 2786 CrossRef CAS PubMed.
- M. Lintuluoto, C. Yamada and J. M. Lintuluoto, J. Phys. Chem. B, 2017, 121, 7235 CrossRef CAS PubMed.
- (a) L. Rulíšek, K. P. Jensen, K. Lundgren and U. Ryde, J. Comput. Chem., 2006, 27, 1398 CrossRef PubMed; (b) M. Srnec, F. Aquilante, U. Ryde and L. Rulíšek, J. Phys. Chem. B, 2009, 113, 6074 CrossRef CAS PubMed.
- P. J. Hart, M. M. Balbirnie, N. L. Ogihara, A. M. Nersissian, M. S. Weiss, J. S. Valentine and D. Eisenberg, Biochemistry, 1999, 38, 2167 CrossRef CAS PubMed.
- I. A. Abreu and D. E. Cabelli, Biochim. Biophys. Acta, 2010, 1804, 263 CrossRef CAS PubMed.
- D. Chen, Q. Wang, H. Zhang, S. Mi, J. Wang, Q. Zeng and G. Zhang, Int. J. Quantum Chem., 2010, 110, 1394 CAS.
- L. Yang, D. R. Powell and R. P. Houser, Dalton Trans., 2007, 955 RSC.
- E. Sperotto, G. P. M. van Klink, J. G. de Vries and G. van Koten, Tetrahedron, 2010, 66, 3478 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1982975. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0cc01926g |
| This journal is © The Royal Society of Chemistry 2020 |