
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceElectronic origin of negative thermal expansion in V2OPO4†
Elise
Pachoud
a,
James
Cumby
a,
Jon
Wright
b,
Branimir
Raguž
c,
Robert
Glaum
c and
J. Paul
Attfield
 *a
*a
aCentre for Science at Extreme Conditions & School of Chemistry, University of Edinburgh, UK. E-mail: j.p.attfield@ed.ac.uk
bEuropean Synchrotron Radiation Facility, Grenoble, France
cInstitut für Anorganische Chemie, Universität Bonn, Germany
First published on 30th April 2020
Abstract
Negative volume expansion between 620 and 800 K in V2OPO4 is discovered to be of electronic origin due to the charge ordering transition at 605 K. Domain reorientation and coexistence of the low and high temperature phases close to the transition are observed in X-ray diffraction data from single crystals grown by chemical vapour transport.
The unusual phenomenon of negative thermal expansion (NTE) where lattice volume decreases upon heating has two broad classes of intrinsic mechanism; structural NTE in framework materials through transverse phonon motion, and electronic NTE from thermal changes in the interatomic bonding potential that may accompany a metal-to-insulator, magnetic, ferroelectric, orbital or charge ordering transition.1,2 Electron delocalisation increases above the latter transitions leading to a hardening of the bonding potential.
NTE was recently reported above a charge ordering transition at Tco = 605 K in V2OPO4.3 This vanadium oxyphosphate adopts a tetragonal I41/amd structure above the transition with stacking along the cT axis of chains of face-sharing V2.5+O6 octahedra running alternately along aT and bT. A monoclinic distortion to C2/c symmetry is observed below Tco with long range V2+/V3+ charge order along the chains. Both structures are shown in Fig. 1. The charge order has been confirmed by X-ray absorption spectroscopy and hard X-ray photoemission spectroscopy studies,4,5 and results in ferrimagnetic order of antiparallel V2+ and V3+ spins below a separate magnetic transition at TC = 164 K. V2OPO4 is insulating and no metal-insulator transition is observed at TC or Tco. Above Tco, an observed reduction of the V–V distance within chains with increasing temperature resulted in overall volume NTE up to the highest studied temperature of 750 K.3
 | ||
| Fig. 1 Crystal structure of V2OPO4; (a) low temperature monoclinic C2/c form showing V2+/V3+ charge order, and (b) high temperature tetragonal I41/amd form. | ||
The tetragonal to monoclinic structural transition has subsequently been confirmed by a single crystal X-ray diffraction study,6 and ab initio electronic structure calculations were recently used to analyse the low temperature ferrimagnetism.7 However, the underlying mechanism for the NTE remains unclear as both a structural possibility, involving loss of V–V bonding interactions at Tco that would enhance transverse oxygen vibrations in the tetragonal phase, and an electronic mechanism resulting directly from loss of V2+/V3+ charge order were proposed.3 Here we report further powder neutron diffraction results and also growth and X-ray analysis of single crystals of V2OPO4, giving insights into the origin of the NTE.
The powder V2OPO4 sample was the same as reported in a previous study.3 High temperature neutron powder diffraction (NPD) data were recorded on the HRPD beamline at the ISIS facility. The sample was placed in a vanadium can under dynamic vacuum during the measurements. Data were collected upon warming from room temperature to 1173 K. Rietveld refinements were carried out with the Fullprof software.8
Single crystals were grown by chemical vapour transport with β-VOPO4 and V metal as starting materials.9 β-VOPO4 was obtained by solution combustion synthesis, reacting a stoichiometric mixture of NH4VO3 and (NH4)2HPO4 in a nitric acid![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :glycine solution with a 3:1 ratio of glycine to vanadium. The solution was dried to a gel over a hot plate. This gel was ignited in a preheated box furnace at 500 °C and kept there for 10 min. The obtained powder was heated successively to 600 °C and 700 °C for 1 day at each temperature with intermediate grinding.10 0.75 g of a mixture of β-VOPO4:V in ratio 1.0:1.1 was placed in a quartz tube with 100 mg of I2 as transport agent. The tube was sealed under vacuum and placed in a two-zone tubular furnace. The temperatures were set at 1000 °C at the source (sample end) and 900 °C at the sink (deposition end). A preliminary heating with reversed temperatures was carried out to clean the growth zone of the ampoule. After 6 days the tube was quenched to room temperature and millimetre-size crystals were obtained.
:glycine solution with a 3:1 ratio of glycine to vanadium. The solution was dried to a gel over a hot plate. This gel was ignited in a preheated box furnace at 500 °C and kept there for 10 min. The obtained powder was heated successively to 600 °C and 700 °C for 1 day at each temperature with intermediate grinding.10 0.75 g of a mixture of β-VOPO4:V in ratio 1.0:1.1 was placed in a quartz tube with 100 mg of I2 as transport agent. The tube was sealed under vacuum and placed in a two-zone tubular furnace. The temperatures were set at 1000 °C at the source (sample end) and 900 °C at the sink (deposition end). A preliminary heating with reversed temperatures was carried out to clean the growth zone of the ampoule. After 6 days the tube was quenched to room temperature and millimetre-size crystals were obtained.
High temperature single crystal X-ray diffraction data were measured on the ID11 beamline at the ESRF synchrotron with λ = 0.189721 Å. A V2OPO4 crystal was cemented with Cerastil on top of a 3 mm quartz capillary. The assembly was then introduced into a 7 mm quartz capillary and sealed under vacuum, previously backfilled twice with argon. Omega scans were collected from room temperature to 1273 K, on warming in 25 K steps up to 873 K and in 100 K steps above, and on cooling in 100 K steps. Temperature was controlled with a hot air blower and measured with a thermocouple on the opposite side of the capillary. Data reduction and integration were performed with the CrysalisPro suite.11
The crystal structure of V2OPO4 was refined against the NPD data in space group C2/c below Tco = 605 K, and in I41/amd above. The monoclinic parameters were transformed to the tetragonal cell setting for comparison purposes, as done in the previous study.3 They are plotted in Fig. 2 together with thermal expansion coefficients, calculated as αl = Δl/lΔT from the tetragonal cell lengths l = aT or cT. The new data agree with the results previously reported up to 750 K, and further reveal the thermal expansion behaviour up to 1173 K. The negative expansion in aT and the resulting NTE in the volume coefficient αV is observed to extend only from 620 up to ∼800 K, with a normal positive thermal expansion (PTE) from 800 to 1200 K. However, no further phase transition is apparent around 800 K as the structure remains tetragonal I41/amd above and below this temperature. Fig. 3 displays the thermal evolution of the tetragonal (204) peak, which is split into monoclinic (−2 2 3) and (2 2 1) below Tco in both neutron and the previously reported powder X-ray data.3 There are no apparent intermediate phases and no changes in width or shape of the tetragonal (204) neutron diffraction peak from 673 and 773 K in the NTE region to 873 K in the PTE regime.
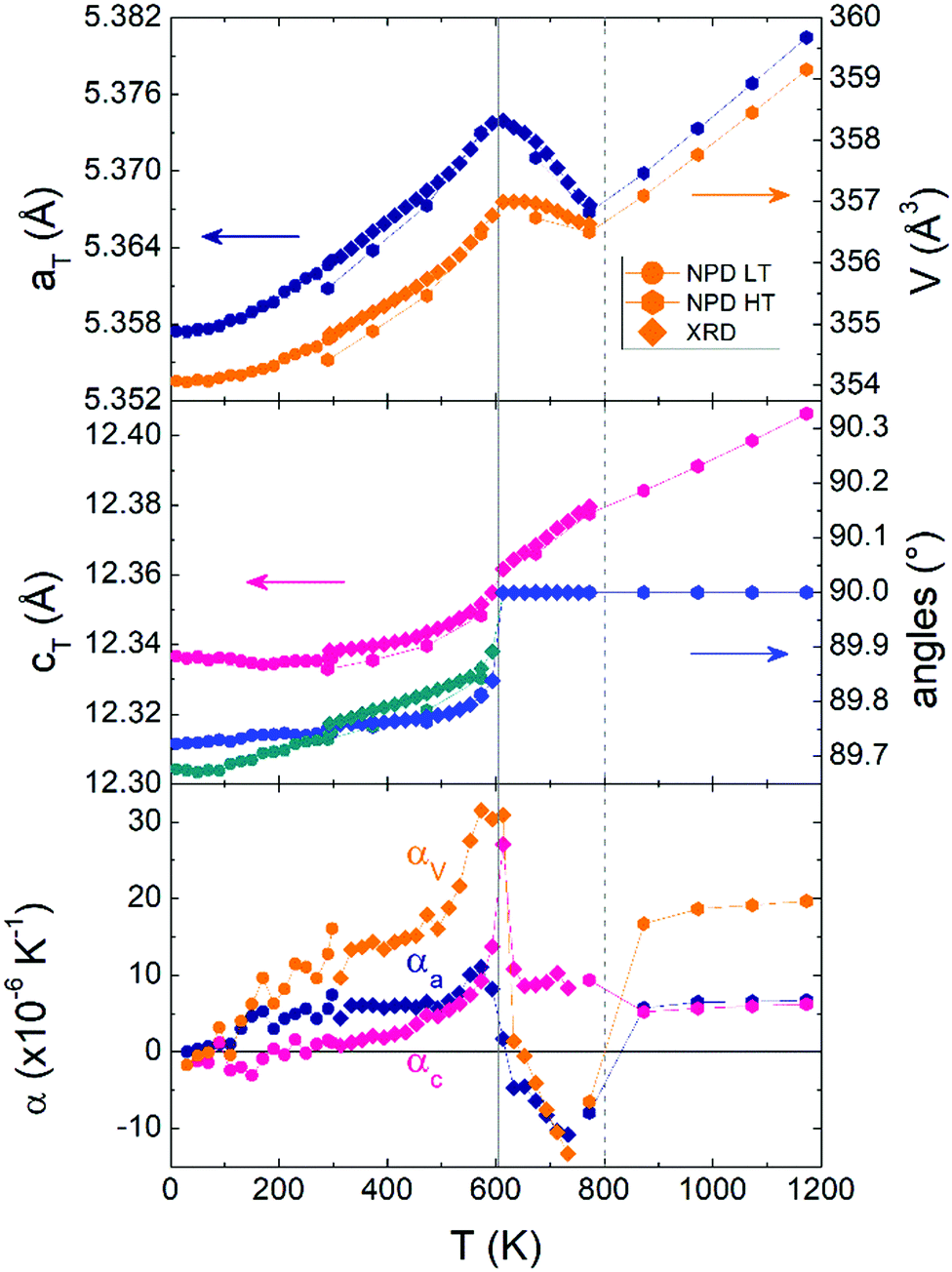 | ||
| Fig. 2 Thermal variations of pseudo-tetragonal unit cell parameters for V2OPO4 and the corresponding thermal expansion coefficients. Low temperature (LT) NPD and powder X-ray diffraction (XRD) data from ref. 3 are shown together with those from the present high temperature (HT) NPD study. | ||
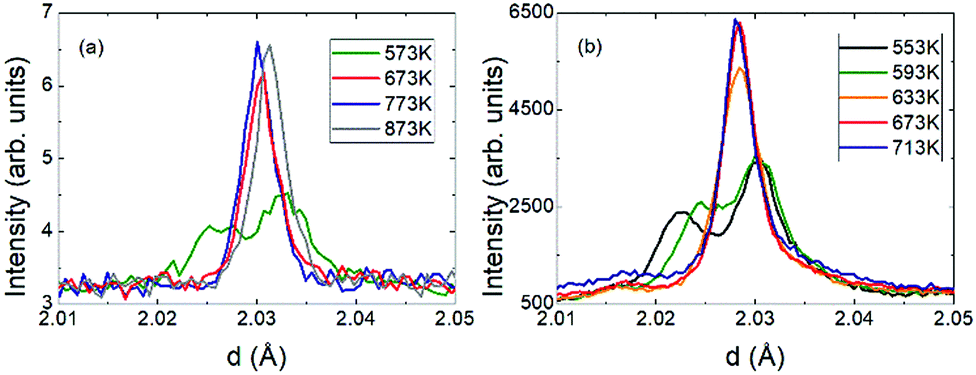 | ||
| Fig. 3 Evolution through the transition of the monoclinic (−2 2 3)/(2 2 1) to the tetragonal (2 0 4) peaks from (a) powder neutron and (b) X-ray diffraction.3 | ||
Anisotropic thermal vibration parameters for the oxygen atoms in the tetragonal phase were refined against powder neutron data from 673 to 973 K and a representative structure (773 K) is shown in Fig. SI-1 (ESI†). The displacement ellipsoid of the phosphate oxygen (site O1, 16h Wyckoff position) is elongated normal to the P–O bond and shortened in the direction of the VO6 chains, while the bridging non-phosphate oxygen (O2, position 4a) is flattened in the direction of cT. No clear discontinuity in the O2 U-factor is observed around the 800 K NTE–PTE change (values are in Table SI-1, ESI†). This, and the absence of a further phase transition around 800 K, tend to rule out a phonon-driven structural mechanism for the NTE but further NPD T-points and vibrational spectra would be needed to definitively rule out a phonon contribution.
Single crystal X-ray diffraction data collected at temperatures up to 548 K were indexed straightforwardly with the C2/c monoclinic cell and those collected above 698 K were unambiguously indexed in tetragonal I41/amd. However, datasets collected between 573 and 673 K showed additional Bragg scattering and the integration process was not straightforward so reliable cell parameters and data for structure refinement were not obtained. We have calculated precession images for these data based on tetragonal indexation (and refinement of the instrument model) from the high temperature cell. A second series of peaks is clearly visible in the calculated (h −1 l) images (Fig. 4a and Fig. SI-2, ESI†), and this reveals a more complex phase behaviour in the vicinity of the charge ordering transition. These additional peaks disappear above 673 K but reappear upon thermal cycling as shown in Fig. SI-3 (ESI†).
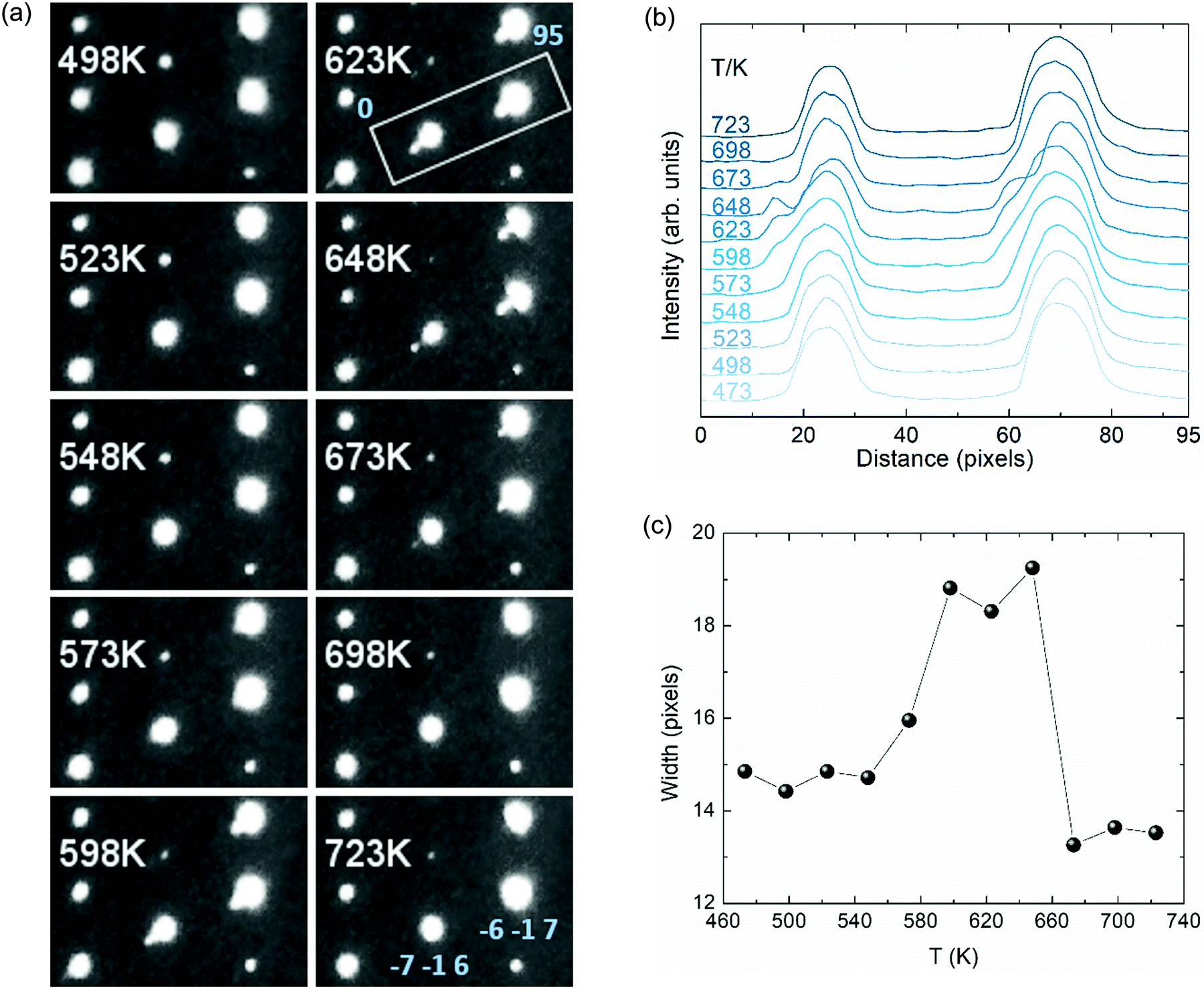 | ||
| Fig. 4 (a) Tetragonal (h −1 l) precession images from high-resolution single crystal X-ray diffraction. (b) Profile plot of the integrated intensity across the highlighted area in the [1 0 1] direction covering the (−7 −1 6) and (−6 −1 7) reflections. (c) FW15%M peak width for the (−7 −1 6) reflection envelope. | ||
To explore the structural evolution of the V2OPO4 crystal through the charge ordering transition, we have extracted intensity profile plots through the tetragonal (−7 −1 6) and (−6 −1 7) reflections as described in the caption to Fig. 4. Additional peaks are visible to the left side of the main peaks in Fig. 4b. These are not in the positions for either the low-temperature monoclinic or high-temperature tetragonal cells, and most likely correspond to formation of an alternative monoclinic domain orientation in part of the crystal, arising through thermal fluctuations as the monoclinic lattice strain decreases. Formation of an intermediate symmetry phase is ruled out by the absence of any additional peaks in the powder diffraction patterns in Fig. 3, however powder data are not sensitive to formation of additional monoclinic domains. The thermal variation of FW15%M (full width at 15% of the maximum intensity above background) for the envelope of the (−7 −1 6) reflection and the additional peak is displayed in Fig. 4c. This demonstrates that the additional domain is present from approximately 560 to 670 K. This lies centrally within the 500 to 700 K region where the volume coefficient αV initially increases on approaching Tco = 605 K and then falls to the most negative value, and so reveals that structural changes accompanying the charge ordering are directly linked to the changes in thermal expansion coefficient.
The above results clearly demonstrate that NTE in V2OPO4 is of electronic origin and is driven by the crossover between a larger-volume low-T charge ordered phase and a smaller-volume high-T phase in which charges are disordered or delocalised. This may be because Tco is the lower limit of a first order transition, or the monoclinic domains may be critical structural fluctuations within the tetragonal bulk. The two phases have intrinsic PTE behaviour, as shown by their respective thermal expansions below 500 and above 800 K. However the volume difference between them leads to NTE above the transition as they interconvert. On cooling from high temperatures, local charge fluctuations lead to the anomalous increase in volume below ∼800 K and αV falls to a minimum value of −13.3 × 10−6 K−1 at 723 K. Initial crystallisation of charge ordered monoclinic domains occurs near this temperature (as observed in the single crystal data), and αV increases towards a maximum of +30 × 10−6 K−1 close to Tco = 605 K. Loss of any residual tetragonal and misoriented monoclinic domains leads αV to fall back to around +15 × 10−6 K−1 on cooling further to 500 K, which represents the background phonon-driven thermal expansion coefficient extrapolated between the regions below (200–500 K) and above (800–1200 K) the electronic transition.
The observation of NTE above Tco in V2OPO4 is similar to observations of NTE over ranges of 50 to 130 K around charge transfer and ordering transitions in LaCu3Fe4O12 (LaCu3+3Fe3+4O12 → LaCu2+3Fe3.75+4O12)12 and Bi0.95La0.05NiO3 (based on Bi3+0.5Bi5+0.5Ni2+O3 → Bi3+Ni3+O3 in the parent material).13 In those cases, much greater (colossal) NTE effects are observed as the charge transfer between cations of different metals is accompanied by an insulator-to-metal transition. In contrast V2OPO4 remains semiconducting throughout the transition and so exemplifies NTE arising purely from loss of charge ordering (V2+V3+OPO4 → V2.5+2OPO4). The overall volume change ΔV/V = −0.3% arises from the average size of ordered V2+ and V3+ in the monoclinic phase being slightly greater than that of the disordered or delocalised V2.5+ state in the tetragonal structure at a given temperature. The structure of V2OPO4 gives rise to highly anisotropic thermal expansion, with NTE only in the aTbT-planes containing the face-sharing chains of VO6 octahedra (αV = −13.3 × 10−6 K−1 at 723 K comes from αa = −10.8 × 10−6 and αc = +8.3 × 10−6 K−1 contributions). Ceramic samples of materials with highly anisotropic thermal expansion such as Ca2RuO414 can show enhanced, microstructural NTE effects,2 and it would be interesting to explore this possibility for V2OPO4.
We thank Drs Alexandra Gibbs and Ka Hou (Jacky) Hong for assistance with facility data collection and ERC, EPSRC and STFC for support.
Conflicts of interest
There are no conflicts to declare.Notes and references
- J. Chen, L. Hu, J. Deng and X. Xing, Chem. Soc. Rev., 2015, 44, 3522 RSC.
- J. P. Attfield, Front. Chem., 2018, 6, 371 CrossRef PubMed.
- E. Pachoud, J. Cumby, C. T. Lithgow and J. P. Attfield, J. Am. Chem. Soc., 2018, 140, 636 CrossRef CAS PubMed.
- K. Murota, E. Pachoud, J. P. Attfield, R. Glaum, R. Sutarto, K. Takubo, D. I. Khomskii and T. Mizokawa, 2020, arXiv, preprint, arXiv:2002.00337.
- K. Murota, E. Pachoud, J. P. Attfield, R. Glaum, T. Yasuda, D. Ootsuki, Y. Takagi, A. Yasui and D. I. Khomskii, T. Mizokawa, in preparation.
- J. Xing, H. Cao, A. Paul, C. Hu, H.-H. Wang, Y. Luo, R. Chaklashiya, J. M. Allred, S. Brown, T. Birol and N. Ni, 2017, arXiv, preprint, arXiv:1712.09973.
- S.-J. Kim and K.-W. Lee, 2019, arXiv, preprint, arXiv:1902.10359.
- J. Rodríguez-Carvajal, Phys. B, 1993, 192, 55 CrossRef.
- R. Glaum and R. Gruehn, Z. Kristall., 1989, 186, 91 Search PubMed.
- S. C. Roy, R. Glaum, D. Abdullin, O. Schiemann, N. Q. Bac and K.-H. Lii, Z. Anorg. Allg. Chem., 2014, 640, 1876 CrossRef CAS.
- Agilent, CrysAlis PRO. Agilent Technologies Ltd, Yarnton, Oxfordshire, England, 2014 Search PubMed.
- Y. W. Long, N. Hayashi, T. Saito, M. Azuma, S. Muranaka and Y. Shimakawa, Nature, 2009, 458, 60 CrossRef CAS PubMed.
- M. Azuma, W.-T. Chen, H. Seki, M. Czapski, S. Olga, K. Oka, M. Mizumaki, T. Watanuki, N. Ishimatsu, N. Kawamura, S. Ishiwata, M. G. Tucker, Y. Shimakawa and J. P. Attfield, Nat. Commun., 2011, 2, 347 CrossRef PubMed.
- K. Takenaka, Y. Okamoto, T. Shinoda, N. Katayama and Y. Sakai, Nat. Commun., 2017, 8, 14102 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0cc01920h |
| This journal is © The Royal Society of Chemistry 2020 |