
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePhthalocyanine–DNA origami complexes with enhanced stability and optical properties†
Ahmed
Shaukat
 a,
Eduardo
Anaya-Plaza
a,
Sofia
Julin
a,
Veikko
Linko
ab,
Tomas
Torres
*cde,
Andrés
de la Escosura
*cd and
Mauri A.
Kostiainen
*ab
a,
Eduardo
Anaya-Plaza
a,
Sofia
Julin
a,
Veikko
Linko
ab,
Tomas
Torres
*cde,
Andrés
de la Escosura
*cd and
Mauri A.
Kostiainen
*ab
aBiohybrid Materials, Department of Bioproducts and Biosystems, Aalto University, FI-00076 Aalto, Finland. E-mail: mauri.kostiainen@aalto.fi
bHYBER Centre, Department of Applied Physics, Aalto University, FI-00076 Aalto, Finland
cDepartment of Organic Chemistry, Universidad Autónoma de Madrid (UAM), Calle Francisco Tomás y Valiente, 7, 28049 Madrid, Spain. E-mail: tomas.torres@uam.es; andres.delaescosura@uam.es
dInstitute for Advanced Research in Chemical Sciences (IAdChem), 28049 Cantoblanco, Madrid, Spain
eIMDEA Nanosience, Ciudad Universitaria de Cantoblanco, 28049 Madrid, Spain
First published on 27th May 2020
Abstract
In this communication, electrostatically assembled phthalocyanine (Pc)–DNA origami (DO) complexes are formed and their optical properties are demonstrated. The formation of the complex prevents the Pc aggregation, thus yielding an enhanced optical response and photooxidative resilience towards aggregation in biologically relevant media. Simultaneously, the Pc protects the DO against enzymatic digestion. Both features solve previous drawbacks associated with phthalocyanine photosensitizers and DNA nanocarriers. The studied complexes may find use in technologies related to the photogeneration of singlet oxygen, e.g., photocatalysis, diagnositic arrays and photodynamic therapy.
DNA origami (DO) is a programmable nucleotide-based nanostructure resulting from the folding of a long, single-stranded DNA (ssDNA) with the help of a custom set of short ssDNA strands.1 The DO method has emerged as a highly versatile tool that opens up a plethora of applications at the interface of biological systems. These 2D and 3D DO2 can carry out predefined tasks, such as controlled drug delivery, targeting and release of cargo.3–7 However, DNA – albeit otherwise being a biocompatible molecule – can be digested or denatured under challenging conditions. For example, foreign DNA from pathogens and damaged endogenous DNA are detected and degraded via complex pathways.8
Overall, the enzymatic digestion of DO is superstructure-dependent.9 Numerous in vitro studies conducted on the stability of DO have revealed that they are more stable than native DNA of similar size.6,10,11 Still, DNA nanostructure-based drug-delivery systems often require modifications to resist, escape or delay the natural defense mechanisms. Several coating strategies to increase stability of the DO have been developed12 such as encapsulation of DO with proteins of viral capsids, serum albumin, and protein–polymer conjugates.13–18 Other coating approaches include e.g. polyethylene glycol (PEG), chitosan, polyethylenimine and other cationic polymers.18–24 These protection strategies have endowed important attributes to DO such as increased resistance against enzymatic degradation13,14,20–23 and reduced immunogenic response.13,23 However, the use of functional dye molecules for DO coating has remained limited.
Photosensitizers (PS) are molecules that absorb light and transfer the energy to an acceptor molecule, normally molecular oxygen present in the surrounding media, generating singlet oxygen (1O2) and other reactive oxygen species (ROS).25–27 Phthalocyanine (Pc) dyes are among the most important porphyrinoid PS, presenting several advantages such as high extinction coefficients, absorption in the red/near-infrared region and high stability.28–32 However, due to the hydrophobic nature of the Pc macrocyclic core, they have a tendency to aggregate in aqueous media, thus decreasing or entirely preventing their photochemical activity. One strategy to circumvent this obstacle is to conjugate Pcs with different biomolecules,33–36 such as DNA, which gives additional advantages including targeting possibilities and enhanced biocompatibility.
To the extent of our knowledge, hybrid materials based on merging DO with functional Pc photosensitizers have not been reported. Morevoer, synergetic materials with resilient photoactivity in biological media simultaneously enhancing the DO stability against enzymatic digestion, has not been achieved. This communication reports a straightforward, non-covalent approach to build up such hybrids, through the electrostatic recognition and complex formation between a positively charged ZnPc (1) and a negatively charged 6-helix bundle (6HB)37 DO (Fig. 1a, for preparation see ESI†). These complexes present maintained, and even enhanced photoactivity in elevated ionic strength media, as well as protection against enzymatic digestion.
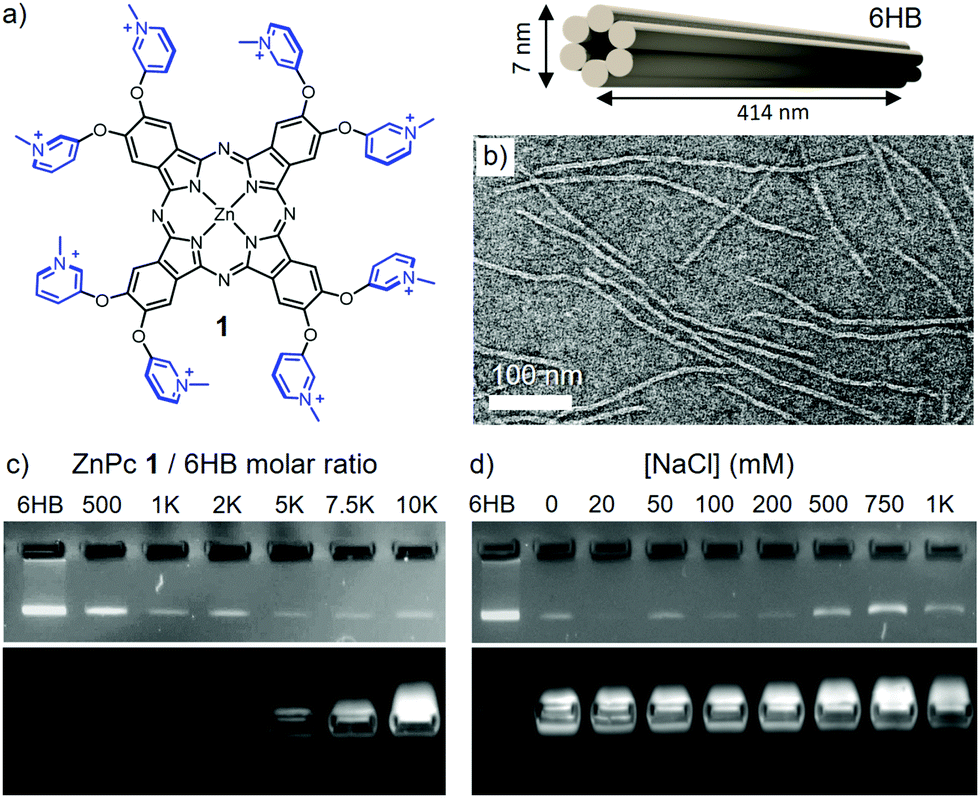 | ||
| Fig. 1 (a) Chemical structure of 1 (left) and schematic representation of 6HB (right). Iodide counterions are not included into the chemical structure for clarity. (b) TEM image of negatively stained 6HB. (c) Agarose gel EMSA of 6HB (4 nM) titrated with increasing molar equivalents of 1. (d) Agarose gel EMSA of the 1–6HB complex (33.75 μM and 4.5 nM, respectively) at increasing NaCl concentrations. The EMSA gels show the EtBr (top) and ZnPc (bottom) emission channels recorded at 532 and 633 nm, respectively. | ||
Transmission electron microscopy (TEM) images of 6HB shows thread-like structures of ∼410 nm in length and 6–7 nm in diameter (Fig. 1b). Mixing of 6HB and compound 1 leads to complex formation, which can be observed as a mesh-like blue network (see Fig. S2, ESI†). The stoichiometric ratio between the components required for complex formation was determined by agarose gel electrophoretic mobility shift assay (EMSA). A constant concentration of 6HB (4 nM) was titrated with 1 in order to determine the maximum amount of Pc that can be grafted on the DO, i.e. the saturation ratio (Fig. 1c). At increasing amounts of 1, the intensity of the free DO band decreases gradually, until saturation at a 1/6HB molar ratio of ca. 2500 eq. (see Fig. S3, ESI†).
In order to verify that the assembly is electrostatically driven, the optical properties and structure morphology of the formed complexes were characterized at different NaCl concentrations.38 To this end, a 1/6HB ratio of 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 eq. was selected, in order to ensure total coverage of the DO. The addition of 50 mM NaCl resulted in the re-dispersion of the blue mesh-like network (see Fig. S5, ESI†), but EMSA still implied that 1 remains bound to the DO up to 200 mM of NaCl (Fig. 1d). Increasing NaCl concentration above 500 mM hinders the complex formation, as indicated by reappearance of the intense 6HB band.
000 eq. was selected, in order to ensure total coverage of the DO. The addition of 50 mM NaCl resulted in the re-dispersion of the blue mesh-like network (see Fig. S5, ESI†), but EMSA still implied that 1 remains bound to the DO up to 200 mM of NaCl (Fig. 1d). Increasing NaCl concentration above 500 mM hinders the complex formation, as indicated by reappearance of the intense 6HB band.
UV-Vis absorption spectroscopy and TEM imaging were utilized to obtain further insights into the relationship between the complex properties and the ionic strength of the media (Fig. 2). The absorption spectrum of 1 at a constant concentration of 2 μM was recorded at 0, 50, 100, 200, 500 and 1000 mM of NaCl, in the presence of 0, 0.5, 1, 2 and 4 nM of 6HB (Fig. 2a and Fig. S6, ESI†). Like for many other ZnPcs, compound 1 is known for a shift in its Q-band absorption wavelength upon aggregation, showing an intense band at 676 nm in the monomeric state, and a broader and lower absorption at 632 nm in the aggregated state.28 Therefore, the ratio between the absorption at 676 and 632 nm determined for different 6HB concentrations indicates the ability of 6HB to prevent the aggregation of 1 (Fig. 2b). In the absence of 6HB, an increase in the ionic strength resulted in the expected aggregation (Fig. 2b, red). However, in the presence of increasing amounts of 6HB, two different effects were observed (Fig. 2b, purple). First, aggregation of 1 is reduced between 0 and 200 mM (regime A), followed by an increase of aggregation between 200 and 1000 mM of NaCl (regime C) (Fig. 2c). The disruption of aggregation is most prominent at a 200 mM NaCl concentration (regime B) and only present at ZnPc/6HB ratios below saturation (i.e. 1 nM of 6HB = 2000 eq. of 1). Thus, the disaggregation effect is a consequence of the complex formation.
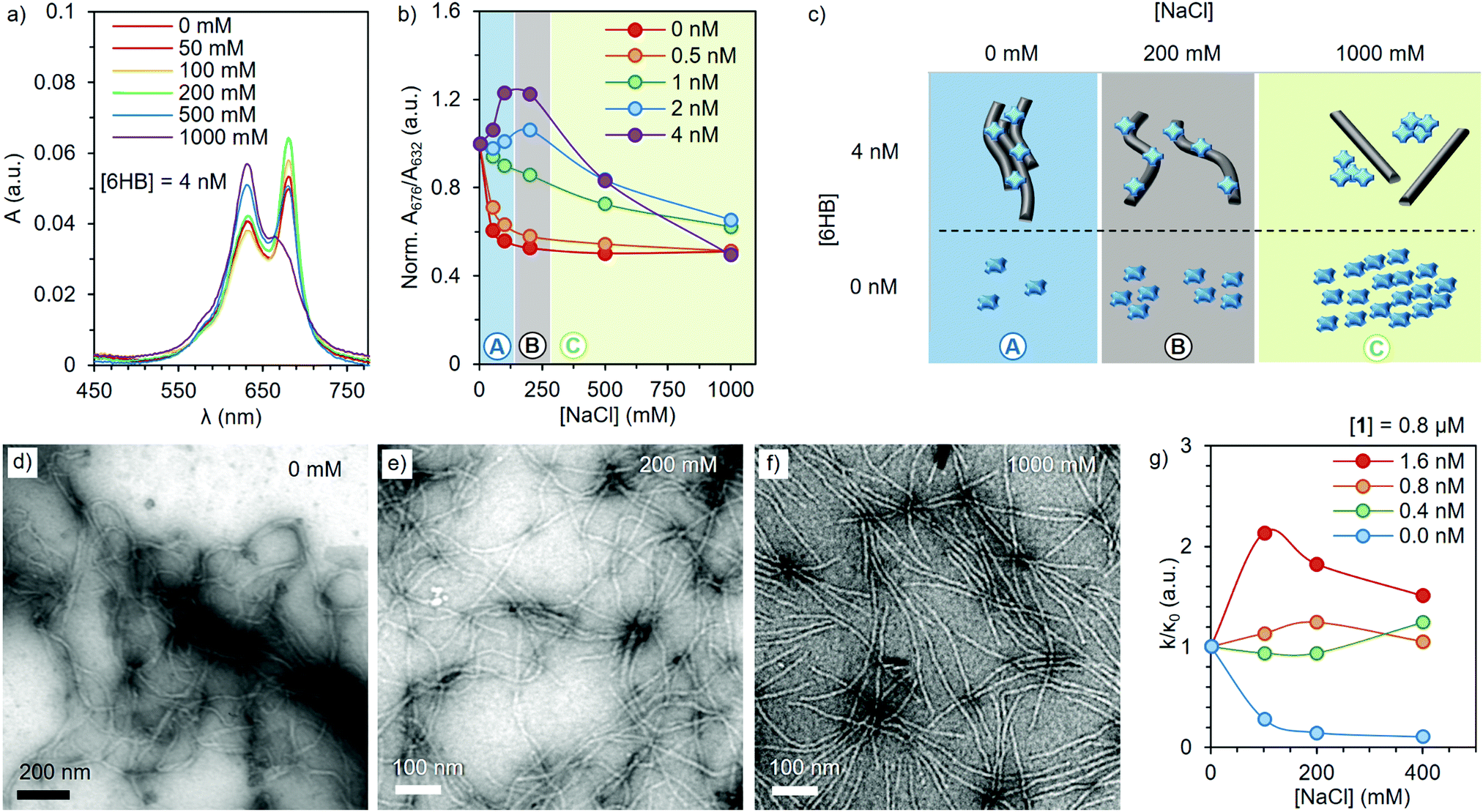 | ||
| Fig. 2 (a) Absorption spectra of 1 (2 μM) in presence of 4 nM of 6HB (1/6HB = 500) at increasing amounts of NaCl. (b) A normalized ratio between the absorption at 676 and 632 nm of 2 μM of 1 at different NaCl and 6HB concentrations (normalized to the ratio at 0 NaCl). (c) Schematic representation of the NaCl effect on the complex (top) and free ZnPc (bottom). Grey rods and blue crosses represent the 6HB and 1, respectively. (d–f) TEM images of the 1–6HB complexes in the presence of 0, 200 and 1000 mM of NaCl, respectively. Samples were stained with uranyl formate (2% w/v) before imaging. (g) Normalized photooxidative activity (k/κ0) of 1–6HB complexes at different 6HB ratios and NaCl concentrations. | ||
TEM images (Fig. 2d–f and Fig. S7, ESI†) verify the change in morphology of the complexes upon an increase of the ionic strength. At low ionic strengths, large bundles of 6HB are found, (also visually observed), as the charged ZnPc serves as a supramolecular glue that crosslinks DO. Subsequent addition of NaCl breaks down the large bundles and single 6HB structures can be observed at [NaCl] = 200 mM (Fig. 2e). Further addition of NaCl results in the detachment of 1 and 6HB (Fig. 1c), with the free ZnPc forming aggregates in the high ionic strength medium. Thus, the formulation of the complexes at 200 mM of NaCl is optimal for obtaining individual DO loaded with high amount of non-aggregated ZnPc, ensuring the ideal optical properties of the complex.
Resilience of the photooxidative properties of the complex towards aggregation induced in physiological conditions, was assessed by the relative method.39 This is based on the direct proportionality between the degradation of a chemical scavenger (1,3-diphenylisobenzofuran, DPBF) and the photogeneration of 1O2 (see ESI† for further details). In order to achieve DPBF solubility in aqueous media, while avoiding any disruptive interaction with the complex (Fig. S8, ESI†), a neutral surfactant (Chremophor EL®) was employed at 0.5% (w/w).35 Briefly, a selection of 1–6HB complexes, with a constant 1 concentration of 0.8 μM and 1/6HB ratios ranging from 500 to 2000 equivalents, were irradiated for different times at constant fluence rates, in the presence of 0, 100, 200 and 400 mM of NaCl and aqueous DPBF (Fig. S9, ESI†). Under these conditions, the ln(A0/At) vs. the irradiation time (with A0 and At being the respective scavenger absorbance values at 417 nm at different irradiation intervals) shows a linear behaviour for all complex ratios, where the slope (k) is related to their capacity for 1O2 generation (Fig. S10, ESI†). Interestingly, plotting the k/κ0, i.e. the ratio between k in the presence of NaCl (Fig. 2g) and k without salt (κ0), reveals that the photoactivity of 1 is strongly quenched at high NaCl concentrations (blue). In contrast, the 1–6HB complex shows an activity maximum between 100 and 200 mM of NaCl (red). These results are in perfect agreement with the aggregation/deaggregation behavior observed previously, and demonstrate that the present complexes can generate 1O2 efficiently in conditions that resemble those of physiological media. Moreover, in these conditions, 6HB showed no evident photodamage,40 rendering a sturdy nanocarrier (Fig. S12, ESI†).
One of the main factors that affect the circulation time and bioavailability of DNA-based drug delivery systems is their susceptibility to enzymatic digestion.23 Deoxyribonuclease (DNase I) is an endonuclease, which degrades DNA into short oligonucleotide fragments.41 In our experiments, 6HB was exposed to increasing amounts of DNase I with varying time spans, and its degradation was monitored by EMSA. A shift in the band to higher mobility was observed, due to degradation of the 6HB structure (d6HB). The enzyme concentration and digestion time were optimized to 80 Kunitz U mL−1 for 30 min (see Fig. S13a, ESI†). However, as the ionic strength of the media affects the enzymatic activity,42 the digestion experiments with 200 mM NaCl were carried out using 360 Kunitz U mL−1 for 180 min (see Fig. S13b, ESI†). Note that these concentrations are well above the biological DNase I concentration (0.256–1.024 Kunitz U mL−1 in 10% FBS).43
In order to evaluate how the coating with 1 protects the 6HBs from DNase I degradation, the following experiment was designed and assessed by EMSA (Fig. 3b and c). The 6HB (2) was subjected to the optimized DNase I digestion resulting in fully digested 6HB (d6HB), which was observed as the disappearance of the DO band and the appearance of fragments with a higher electrophoretic mobility (3). The 1–6HB complex was formed by the addition of 4000 eq. of ZnPc, in order to avoid undesired interaction between the enzyme and the excess of PS in solution (4). However, both the intact 6HB and the digestion products can bind to 1 and have similar electrophoretic mobility, and therefore d6HB interaction with 1 was also included as a control (8 and 9). In order to assess the integrity of the DO, the electrostatic assembly was reversed by the addition of 597 mM heparin (800 times higher charge concentration than in 1). This highly anionic biopolymer strongly binds to 1, which release the intact 6HB (5). Finally, 1–6HB was subjected to the same digestion conditions (6) and subsequently treated with heparin, resulting in the recovery of intact 6HB (7). To prove that the DO protection is achieved solely through the interaction with 1, and not due to the macroscopic complexation or aggregation, the same experiment was repeated in the presence of 200 mM NaCl yielding the same result (Fig. 3c, 7).
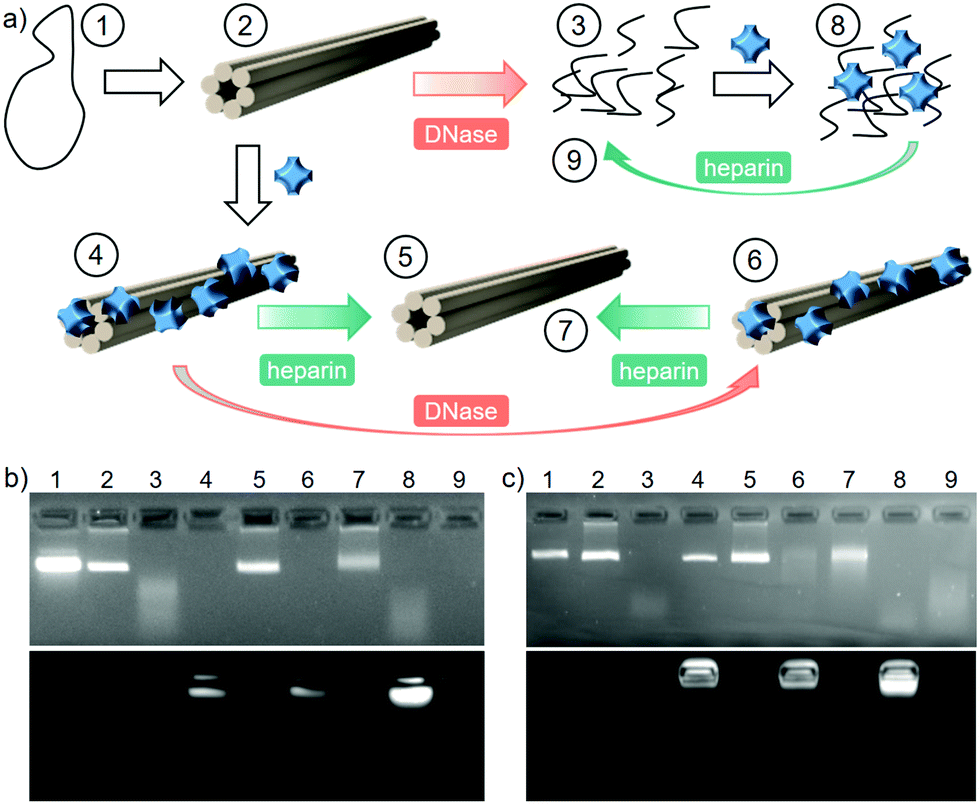 | ||
| Fig. 3 (a) Scheme of the digestion experiment. (1) M13mp18 scaffold, (2) 6HB, (3) d6HB, (4) ZnPc 1–6HB, (5) deprotected 6HB, (6) ZnPc 1–6HB after digestion, (7) deprotected 6HB after digestion, (8) ZnPc 1–d6HB, (9) deprotected d6HB. The numbers in the figure correspond to the agarose gel EMSA lanes at 0 (b) and 200 mM (c). | ||
In conclusion, the electrostatic complexation of 1 and 6HB represents a straightforward and synergistic approach towards the development of DNA-based photosensitizing systems. At optimal conditions, the DO-bound ZnPc is in its non-aggregated state, which retains the PS capacity for 1O2 generation. The DO nanocarrier, in turn, is simultaneously protected against enzymatic digestion, potentially improving its bioavailability. Additionally, fine-tuning of the optical and morphological features of the complex can be achieved through the control over the ionic strength within biologically relevant range and compatible with DO.44 Considering that the photogeneration of 1O2 and other ROS plays a key role in various technologies at the interface with biological systems such as diagnostic arrays, photodynamic therapy and oxidative photocatalysis, this work opens new research avenues based on materials that merge programmable DNA nanostructures and Pc functional dyes.
We acknowledge the Academy of Finland (286845, 308578), the Marie Sklodowska-Curie grant (794536), Emil Aaltonen Foundation, The Swedish Cultural Foundation, Jane and Aatos Erkko Foundation, Sigrid Jusélius Foundation and the Spanish MINECO (CTQ2017-85393-P and CTQ-2017-89539-P, PCIN-2017-042/EuroNanoMed2017-191, TEMPEAT) with European Union FEDER funds. The work was carried out under the Academy of Finland's Centre of Excellence Programme (2014–2019). IMDEA Nanociencia acknowledges support from the “Severo Ochoa” Programme for Centres of Excellence in R&D (MINECO, Grant SEV2016-0686). We acknowledge the provision of facilities and technical support by Aalto University Bioeconomy Facilities and OtaNano – Nanomicroscopy Center (Aalto-NMC).
Conflicts of interest
The authors declare no conflict of interest.Notes and references
- P. W. K. Rothemund, Nature, 2006, 440, 297–302 CrossRef CAS PubMed.
- S. Nummelin, J. Kommeri, M. A. Kostiainen and V. Linko, Adv. Mater., 2018, 30, 1703721 CrossRef PubMed.
- V. Linko, A. Ora and M. A. Kostiainen, Trends Biotechnol., 2015, 33, 586–594 CrossRef CAS PubMed.
- Q. Jiang, S. Liu, J. Liu, Z. Wang and B. Ding, Adv. Mater., 2019, 31, 1804785 CrossRef CAS PubMed.
- C. E. Castro, F. Kilchherr, D.-N. Kim, E. L. Shiao, T. Wauer, P. Wortmann, M. Bathe and H. Dietz, Nat. Methods, 2011, 8, 221–229 CrossRef CAS PubMed.
- P. Zhan, Q. Jiang, Z. Wang, N. Li, H. Yu and B. Ding, ChemMedChem, 2014, 9, 2013–2020 CrossRef CAS PubMed.
- A. Keller and V. Linko, Angew. Chem., Int. Ed., 2020 DOI:10.1002/anie.201916390.
- D. Smith, V. Schüller, C. Engst, J. Rädler and T. Liedl, Nanomedicine, 2013, 8, 105–121 CrossRef CAS PubMed.
- S. Ramakrishnan, B. Shen, M. A. Kostiainen, G. Grundmeier, A. Keller and V. Linko, ChemBioChem, 2019, 20, 2818–2823 CrossRef CAS PubMed.
- Q. Jiang, C. Song, J. Nangreave, X. Liu, L. Lin, D. Qiu, Z.-G. Wang, G. Zou, X. Liang, H. Yan and B. Ding, J. Am. Chem. Soc., 2012, 134, 13396–13403 CrossRef CAS PubMed.
- J.-W. Keum and H. Bermudez, Chem. Commun., 2009, 7036 RSC.
- N. Stephanopoulos, ChemBioChem, 2019, 20, 2191–2197 CrossRef CAS PubMed.
- H. Auvinen, H. Zhang, Nonappa, A. Kopilow, E. H. Niemelä, S. Nummelin, A. Correia, H. A. Santos, V. Linko and M. A. Kostiainen, Adv. Healthcare Mater., 2017, 6, 1700692 CrossRef PubMed.
- N. A. Estrich, A. Hernandez-Garcia, R. de Vries and T. H. LaBean, ACS Nano, 2017, 11, 831–842 CrossRef CAS PubMed.
- A. Lacroix, T. G. W. Edwardson, M. A. Hancock, M. D. Dore and H. F. Sleiman, J. Am. Chem. Soc., 2017, 139, 7355–7362 CrossRef CAS PubMed.
- A. Hernandez-Garcia, N. A. Estrich, M. W. T. Werten, J. R. C. Van Der Maarel, T. H. LaBean, F. A. de Wolf, M. A. Cohen Stuart and R. de Vries, ACS Nano, 2017, 11, 144–152 CrossRef CAS PubMed.
- J. Mikkilä, A.-P. Eskelinen, E. H. Niemelä, V. Linko, M. J. Frilander, P. Törmä and M. A. Kostiainen, Nano Lett., 2014, 14, 2196–2200 CrossRef PubMed.
- S. Ramakrishnan, H. Ijäs, V. Linko and A. Keller, Comput. Struct. Biotechnol. J., 2018, 16, 342–349 CrossRef CAS PubMed.
- J. K. Kiviaho, V. Linko, A. Ora, T. Tiainen, E. Järvihaavisto, J. Mikkilä, H. Tenhu, Nonappa and M. A. Kostiainen, Nanoscale, 2016, 8, 11674–11680 RSC.
- Y. Ahmadi, E. De Llano and I. Barišić, Nanoscale, 2018, 10, 7494–7504 RSC.
- N. P. Agarwal, M. Matthies, F. N. Gür, K. Osada and T. L. Schmidt, Angew. Chem., Int. Ed., 2017, 56, 5460–5464 CrossRef CAS PubMed.
- N. Ponnuswamy, M. M. C. Bastings, B. Nathwani, J. H. Ryu, L. Y. T. Chou, M. Vinther, W. A. Li, F. M. Anastassacos, D. J. Mooney and W. M. Shih, Nat. Commun., 2017, 8, 15654 CrossRef CAS PubMed.
- S. D. Perrault and W. M. Shih, ACS Nano, 2014, 8, 5132–5140 CrossRef CAS PubMed.
- A. Chopra, S. Krishnan and F. C. Simmel, Nano Lett., 2016, 16, 6683–6690 CrossRef CAS PubMed.
- A. P. Castano, P. Mroz and M. R. Hamblin, Nat. Rev. Cancer, 2006, 6, 535–545 CrossRef CAS PubMed.
- Y.-Y. Wang, Y.-C. Liu, H. Sun and D.-S. Guo, Coord. Chem. Rev., 2019, 395, 46–62 CrossRef CAS.
- Q. Sun, M. Barz, B. G. De Geest, M. Diken, W. E. Hennink, F. Kiessling, T. Lammers and Y. Shi, Chem. Soc. Rev., 2019, 48, 351–381 RSC.
- T. Nyokong, Coord. Chem. Rev., 2007, 251, 1707–1722 CrossRef CAS.
- X. Li, B.-D. Zheng, X.-H. Peng, S.-Z. Li, J.-W. Ying, Y. Zhao, J.-D. Huang and J. Yoon, Coord. Chem. Rev., 2019, 379, 147–160 CrossRef CAS.
- R. C. H. Wong, P.-C. Lo and D. K. P. Ng, Coord. Chem. Rev., 2019, 379, 30–46 CrossRef CAS.
- S. Singh, A. Aggarwal, N. V. S. D. K. Bhupathiraju, G. Arianna, K. Tiwari and C. M. Drain, Chem. Rev., 2015, 115, 10261–10306 CrossRef CAS PubMed.
- C. G. Claessens, U. Hahn and T. Torres, Chem. Rec., 2008, 8, 75–97 CrossRef CAS PubMed.
- V. Almeida-Marrero, E. van de Winckel, E. Anaya-Plaza, T. Torres and A. de la Escosura, Chem. Soc. Rev., 2018, 47, 7369–7400 RSC.
- X. Xue, A. Lindstrom and Y. Li, Bioconjugate Chem., 2019, 30, 1585–1603 CrossRef CAS PubMed.
- J. Mikkilä, E. Anaya-Plaza, V. Liljeström, J. R. Caston, T. Torres, A. de la Escosura and M. A. Kostiainen, ACS Nano, 2016, 10, 1565–1571 CrossRef PubMed.
- E. Anaya-Plaza, A. Aljarilla, G. Beaune, Nonappa, J. V. I. Timonen, A. Escosura, T. Torres and M. A. Kostiainen, Adv. Mater., 2019, 31, 1902582 CrossRef PubMed.
- H. Bui, C. Onodera, C. Kidwell, Y. Tan, E. Graugnard, W. Kuang, J. Lee, W. B. Knowlton, B. Yurke and W. L. Hughes, Nano Lett., 2010, 10, 3367–3372 CrossRef CAS PubMed.
- A. M. Smith, A. A. Lee and S. Perkin, J. Phys. Chem. Lett., 2016, 7, 2157–2163 CrossRef CAS PubMed.
- N. Nombona, K. Maduray, E. Antunes, A. Karsten and T. Nyokong, J. Photochem. Photobiol., B, 2012, 107, 35–44 CrossRef CAS PubMed.
- L. Chen, Y. Zhao, X. Sun, J. Jiang, F. Wu and K. Wang, J. Porphyrins Phthalocyanines, 2019, 23, 655–663 CrossRef CAS.
- Z. Zhou, C. Zhu, J. Ren and S. Dong, Anal. Chim. Acta, 2012, 740, 88–92 CrossRef CAS PubMed.
- G. Alzbutas, M. Kaniusaite and A. Lagunavicius, PLoS One, 2016, 11, e0150404 CrossRef PubMed.
- J. Hahn, S. F. J. Wickham, W. M. Shih and S. D. Perrault, ACS Nano, 2014, 8, 8765–8775 CrossRef CAS PubMed.
- C. Kielar, Y. Xin, B. Shen, M. A. Kostiainen, G. Grundmeier, V. Linko and A. Keller, Angew. Chem., Int. Ed., 2018, 57, 9470–9474 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details and methods, supplementary EMSA gels, optical microscopy images, additional TEM images, photoactivity and photostability assays and UV-Vis spectroscopy graphs. See DOI: 10.1039/d0cc01916j |
| This journal is © The Royal Society of Chemistry 2020 |