
Strain release – an old tool for new transformations
 a
Dorota
Gryko
*a
a
Dorota
Gryko
*a
Abstract
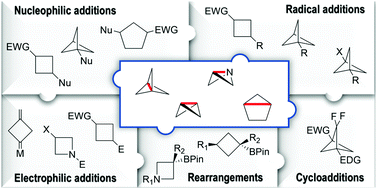
Strain-release driven transformations give access to attractive bioisosteric motifs highly prized by medicinal chemists and they are characteristic of molecules possessing distorted bond lengths and angles. By broadening the chemical space in drug discovery, recently, these compounds have attracted a lot of interest. Their reactivity stems mainly from an increased energy and destabilization. As a result, the opening of the bridging bond occurs under the action of both nucleophiles and electrophiles as well as radical species and transition metals. Though the bridge bond dominates their reactivity, it is also influenced by the substitution pattern. This feature article focuses on strain-release driven strategies paying particular attention to the most recent (year > 2010) advances.
 Please wait while we load your content...
Please wait while we load your content...

