
Valence tautomerism in a cobalt-verdazyl coordination compound†

Abstract
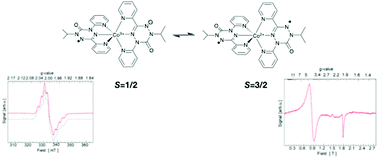
Coordination of 1-isopropyl-3,5-dipyridyl-6-oxoverdazyl to cobalt results in a dication best described in the solid state as a high spin cobalt(II) ion coordinated to two radical ligands with an S = 3/2 ground state. On dissolution in acetonitrile, the cobalt(II) form equilibrates with a cobalt(III) valence tautomer with an S = 1/2 ground state.
 Please wait while we load your content...
Please wait while we load your content...

