
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Enhanced single-molecule magnetism in dysprosium complexes of a pristine cyclobutadienyl ligand†
James P.
Durrant
 ab,
Jinkui
Tang
c,
Akseli
Mansikkamäki
*d and
Richard A.
Layfield
*a
ab,
Jinkui
Tang
c,
Akseli
Mansikkamäki
*d and
Richard A.
Layfield
*a
aDepartment of Chemistry, School of Life Sciences, University of Sussex, Brighton, BN1 9QR, UK. E-mail: R.Layfield@sussex.ac.uk
bSchool of Chemistry, The University of Manchester, Oxford Road, Manchester, M13 9PL, UK
cChangchun Institute of Applied Chemistry, Chinese Academy of Sciences, Renmin Street 5626, 130022 Changchun, China
dNMR Research Unit, University of Oulu, P.O. Box 8000, FI-90014, Finland. E-mail: Akseli.Mansikkamaki@oulu.fi
First published on 19th March 2020
Abstract
Intact transfer of the cyclobutadienyl ligand [C4(SiMe3)4]2− to yttrium and dysprosium (M) produces the half-sandwich complexes [M{η4-C4(SiMe3)4}(BH4)2(THF)]− as coordination polymers with bridging sodium or potassium ions. The dysprosium versions are single-molecule magnets (SMMs) with energy barriers of 371(7) and 357(4) cm−1, respectively. The pristine cyclobutadienyl ligands provide a strong axial crystal field that enhances the SMM properties relative to related cyclopentadienyl compounds.
Single-molecule magnets (SMMs) are coordination compounds in which individual molecules have a bi-stable magnetic ground state.1 SMMs based on lanthanides dominate the landscape,2,3 with terbium and dysprosium being the most popular owing to the strong spin–orbit coupling and magnetic anisotropy of Tb3+ and Dy3+ ions, which are required to observe magnetic hysteresis. Indeed, the properties of Dy3+ are so well-suited to the task that dysprosium compounds not showing SMM traits are almost as remarkable as those that do. Beyond the fundamental interest in SMMs, some key systems have been used as components in prototype molecular spintronic devices, such as spin valves and spin transistors.4,5
Two parameters often used to quantify SMM performance are the effective energy barrier to reversal of the magnetization (Ueff) and the magnetic blocking temperature (TB), which can be defined as the highest temperature at which magnetic hysteresis occurs.6 Whilst SMMs with large Ueff values are not uncommon, examples with blocking temperatures above the liquid-helium regime are rare. Some of us recently reported the metallocene [(C5Me5)Dy(C5iPr5)]+,7 which has a blocking temperature of 80 K, i.e. above the boiling point of liquid nitrogen, and the broader significance of this system is that it provides a blueprint for further improving the performance of dysprosium SMMs through targeted modifications to the chemistry.8 In the case of dysprosium, large Ueff and TB values occur when the crystal field is both strong and highly axial. Some increases in TB beyond 80 K may be achievable with metallocene SMMs by modifying the substituents, yet more profound changes are required to observe major improvements in performance. Therefore, we have recently focused on the cyclobutadienyl (Cb) ligand in f-element chemistry.9–11
The rationale for replacing Cp ligands with Cb in dysprosium SMMs is based on the idea that the greater charge density of the four-membered ring, which is 6π aromatic and therefore dianionic, will produce a stronger (axial) crystal field. As such, (half-)sandwich Cb complexes of dysprosium should have improved SMM properties relative to analogous Cp complexes. To test this, we synthesized two complexes containing the half-sandwich unit {η4-C4(SiMe3)4}Dy by reacting [Dy(BH4)3(THF)3] with the alkali metal cyclobutadienyl salts [A2{C4(SiMe3)4}] (A = Na, K) in benzene according to Scheme 1. The products were identified by X-ray crystallography as the coordination polymers [Dy{η4-C4(SiMe3)4}(BH4)2(THF)Na]∞ (1Dy) and [Dy{η4-C4(SiMe3)4}(BH4)2(THF)K]∞ (2Dy). To broaden the scope of the synthetic method, the analogous yttrium compounds [Y{η4-C4(SiMe3)4}(BH4)2(THF)Na]∞ (1Y) and [Y{η4-C4(SiMe3)4}(BH4)2(THF)K]∞ (2Y) were also synthesized (Tables S1–S3 and Fig. S1–S11, ESI†).
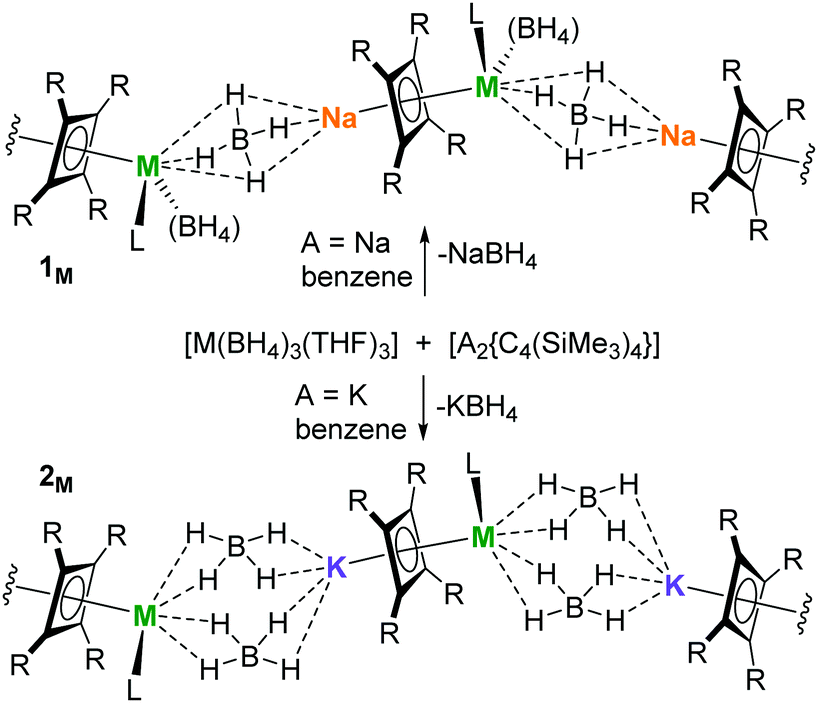 | ||
| Scheme 1 Synthesis of 1M and 2M (M = Y, Dy; R = SiMe3, L = THF). | ||
The structures of 1Y and 1Dy are alike, hence only the dysprosium version is described here (see Fig. S13 for details of 1Y). The structure of 1Dy consists of an asymmetric unit in which two similar yet crystallographically unique half-sandwich units alternate along zigzag polymeric chains (Fig. 1). Each dysprosium centre is ligated by an η4-C4(SiMe3)4 ligand, a bridging borohydride ligand, a terminal κ3-bound borohydride ligand and a THF ligand. The different coordination modes of the borohydride ligands in 1Dy are reflected in the FTIR spectrum, which shows absorptions at 2450 cm−1 and 2100–2300 cm−1 for the terminal and bridging B–H bonds, respectively (Fig. S12, ESI†). From Dy1, the polymer assembles through the Cb ligand in one direction with a μ:η4-interaction with Na1, and in the opposite direction via coordination of the bridging borohydride ligand to Na2. The Dy–C distances and the Dy–Cbcent distance in molecule 1 of 1Dy are 2.489(8)–2.500(8) Å and 2.262(4) Å, respectively, the Na–C and Na–Cbcent distance are 2.591(9)–2.637(9) Å and 2.392(6) Å, respectively, and the Dy–Cb–Na angle is 178.4(2)°. The solution-phase 1H and 13C NMR spectra of 1Y in THF-D8 are consistent with the solid-state structure, as are the 23Na and 29Si NMR spectra, which feature resonances at δ = −4.26 ppm and δ = −22.69 ppm, respectively (Fig. S1–S4, ESI†). The 11B and 11B{1H} spectra both contain broad resonances centred on δ = −23.00 ppm, with unresolved 1J coupling to the protons (Fig. S5 and S6, ESI†), which may point to the borohydride ligands in 1Y undergoing exchange in solution.
 | ||
| Fig. 1 Thermal ellipsoid representations (50% probability) of segments of the polymeric structures of 1Dy (top) and 2Dy (bottom). H = white, B = pink, C = black, O = red, Na = orange, Si = grey, K = lilac, Dy = green. For clarity, only the hydrogen atoms bound to boron are shown. | ||
The structures of 2Y and 2Dy are comparable to those of the sodium analogues, consisting of zigzag chains based on [{η4-C4(SiMe3)4}M(BH4)2(THF)K] units (Fig. 1 and Fig. S14, ESI†). The structural similarities are reflected in the FTIR spectra of all four compounds, which feature absorptions at similar frequencies in the region 450–4000 cm−1 (Fig. S12, ESI†). The only notable difference in the structures of 1M and 2M is that both borohydride ligands in the latter bridge between potassium and the rare earth element owing to the larger radius of potassium relative to sodium. In 2Dy, the Dy–C, Dy–Cbcent, K–C and K–Cbcent distances are 2.473(6)–2.519(6) Å, 2.264(3) Å, 2.890(6)–2.929(6) Å and 2.730(3) Å, respectively, and the Dy–Cb–K angle is 176.46(13)° (details of 2Y are provided in the ESI†). In solution, each 1H and 13C environment in 2Y produces a characteristic resonance in the NMR spectrum (Fig. S7 and S8, ESI†), with the 11B{1H} and 29Si NMR spectra showing resonances at δ = −27.01 ppm and −22.58 ppm, respectively (Fig. S9 and S11, ESI†). The 11B{1H} resonance is sharp and the 1J coupling (84 Hz) to the protons is well resolved as a quintet in the 11B NMR spectrum (Fig. S10, ESI†), suggesting that the borohydride ligands in 2Y are not in exchange.
The cyclobutadienyl chemistry of the f-elements is in its infancy, with the first examples being reported only recently.9–12 In stark contrast to the variety of substituents used with cyclopentadienyl ligands, the scope for substituent variation with cyclobutadienyl ligands is severely restricted owing to the inherent instability of anti-aromatic, strained cyclobutadiene pro-ligands. Currently, the only cyclobutadiene that can be isolated in synthetically useful amounts is the tetrakis(trimethylsilyl) derivative employed in this study.13 However, a drawback of using C4(SiMe3)4 with f-elements is that the methyl substituents are prone to deprotonation, which results in formation of tuck-in complexes, i.e. additional coordination to the metal by a pendant [CbSiMe2CH2]− group.9 Furthermore, the dianionic cyclobutadienyl ring is susceptible to protonation (sometimes from one of its own SiMe3 substituents), leading to loss of aromaticity and formation of η3-cyclobutenyl ligands.10 In light of this, the intact transfer of [C4(SiMe3)4]2− to yttrium and dysprosium in 1M and 2M is noteworthy. Compounds 1Dy and 2Dy therefore provide the first opportunity to study the influence of a pristine cyclobutadienyl ligand on the dynamic magnetic properties of dysprosium.
The magnetic susceptibility of 1Dy and 2Dy was measured in static (DC) and dynamic (AC) fields using a Magnetic Property Measurement System. In a static field of 1000 Oe, the temperature dependence of the molar magnetic susceptibility (χM) for both compounds is typical of Dy3+ with a 6H15/2 ground multiplet (Fig. S15 and S25, ESI†). Thus, χMT at 300 K is 13.46 cm3 K mol−1 and 13.52 cm3 K mol−1 for 1Dy and 2Dy, respectively, with a gradual decrease occurring down to about 9 K, when a sharper decrease is observed and values of 9.60 cm3 K mol−1 and 9.13 cm3 K mol−1, respectively, are reached at 2 K. The SMM properties of the two compounds were then characterized using the frequency-dependence of the imaginary component of the AC susceptibility, χ′′(ν), at various temperatures. In the case of 1Dy, maxima in χ′′(ν) were observed from 1.9 K to 39 K, however up to 9 K the frequency at which the maxima occur is essentially temperature independent (Fig. 2). At higher temperatures, the maxima shift to higher frequencies before reaching the upper limit of the instrument at 1488 Hz. Fits of the AC susceptibility using the standard Cole–Cole method and α-parameters of 0–0.36 allowed the relaxation times (τ) to be extracted (Fig. S17–S22 and Table S4, ESI†). Plotting τ vs. T−1 gave good fits of the data using τ−1 = τ0−1e−Ueff/kBT + CTn + τQTM−1, where the Orbach parameters are τ0−1 and Ueff, the Raman parameters are C and n, and the rate of quantum tunnelling of the magnetization (QTM) is τQTM−1 (Fig. 2). The analysis for 1Dy gave Ueff = 371(7) cm−1 (or 534(10) K) with τ0 = 7.8(2) × 10−12 s, C = 0.0041(7) s−1 K−n, n = 3.85(6) and τQTM = 0.041(1) s. The χ′′(ν) data for 2Dy (Fig. 2 and Fig. S27–S32, Table S5, ESI†) are similar to that of 1Dy, and the same fitting procedure with α = 0–0.31 gave Ueff = 357(4) cm−1 (or 513(6) K), with τ0 = 1.6(3) × 10−10 s, C = 0.014(1) s−1 K−n, n = 2.89(2) and τQTM = 0.35(1) s (Fig. 2).
 | ||
| Fig. 2 Frequency dependence of the out-of-phase susceptibility for 1Dy (top) and 2Dy (middle) in zero DC field; solid lines represent fits to the data using the parameters stated in the ESI.† Bottom: Temperature dependence of the relaxation times for 1Dy (black circles) and 2Dy (blue circles) using the parameters stated in the main text. | ||
The temperature dependence of τ for 1Dy and 2Dy implies that magnetic relaxation is characterized by Orbach processes from 39 K down to about 10 K, and then QTM dominates. A qualitative explanation for the SMM behaviour is that each Dy3+ ion experiences an axial crystal field provided by the cyclobutadienyl ligand, with the THF and borohydride ligands providing a competing equatorial crystal field, as evidenced by the fast QTM at lower temperatures. Support for this idea can obtained upon comparison of 1Dy and 2Dy with the half-sandwich complex [(η5-C5iPr5)Dy(BH4)2(THF)], an SMM with a barrier of 241(7) cm−1 in zero DC field.7 The differing barrier heights in these Cb- and Cp-ligated half-sandwich compounds is presumably related to the stronger axial crystal field provided by the pristine dianionic [C4(SiMe3)4]2− ligand relative to that provided by the monoanionic [C5iPr5]− ligand.
A quantitative description of the electronic structure of 2Dy was obtained by performing multireference ab initio calculations on a model consisting of three [{η4-C4(SiMe3)4}M(BH4)2(THF)]− units and two bridging K+ ions (Fig. 3).14–21 The Dy3+ ions in the two capping half-sandwich units were replaced by Y3+ in order to study only the central Dy3+ ion. The positions of the hydrogen atoms were optimized using density functional theory (DFT) while the positions of heavier atoms were fixed to their crystal-structure coordinates. DFT calculations were carried out to ensure that no significant changes in the charge distribution of the central fragment would be obtained by including a longer fragment of 2Dy.
 | ||
| Fig. 3 Top: The principal magnetic axis of the ground doublet of Dy3+ in 2Dy. Bottom: The qualitative relaxation barrier in 2Dy. The arrows represent transition magnetic moments, with stronger arrows indicating larger values. | ||
The principal magnetic axis of the ground Kramers doublet (KD) is shown in Fig. 3 and the properties of the eight lowest Kramers doublets (KDs) arising from the crystal-field split ground 6H15/2 multiplet of the Dy3+ ion are listed in Table S5. The direction of the principal magnetic axis in the ground KD is clearly set by the strong axial interaction between Dy3+ and the [C4(SiMe3)4]2− ligand. The ground KD has an axial g-tensor with vanishingly small transverse components suggesting that QTM should be slow at zero field. The transverse components become significant in the second excited KD, indicating that the barrier for the relaxation of magnetization is crossed at that point. This gives a predicted effective barrier height of 326 cm−1, which is in good agreement with the experimentally determined value of 357(4) cm−1 for 2Dy. The transverse components are small enough that transitions up to the fourth excited could, in principle, be possible, but this is improbable.
The effective energy barrier was also modeled for 2Dy by calculating the transition magnetic moment matrix elements between the different states in the ground multiplet following a well-established method.20 The results (Fig. 3 and Table S6, ESI†) indicate that the barrier is crossed at the second excited KD. It should be noted that values of the transition magnetic moments remain relatively small up to the fifth excited KD indicating that even minor modifications to the molecular structure, and hence the crystal-field, could significantly increase the barrier height.
Further insight into the electronic structure of 2Dy was obtained by calculating the ab initio crystal-field parameters according to a well-known methodology (Tables S7 and S8, ESI†).21 The axial second-rank parameter is B20 = −377 cm−1, which creates a relatively strong axial field. However, the off-diagonal second-rank parameters also have large magnitudes, i.e. |B2±1| = 15 cm−1 and |B2±2| = 19 cm−1, leading to significant mixing of the different states characterized by a definite projection of the total angular momentum. Compared to [(C5Me5)Dy(C5iPr5)]+,7 the off-diagonal second-rank parameters have very similar magnitudes in 2Dy, but the axial parameters are roughly four times smaller. This means that the axiality in 2Dy, originating from the interaction with the [C4(SiMe3)4]2− ligand, is not strong enough to overcome the significant non-axial components of the crystal field arising from the [BH4]− and THF ligands.
In summary, the main finding in this study confirms our hypothesis that pristine cyclobutadienyl ligands can indeed generate much stronger axial crystal fields at dysprosium centres than more commonly used cyclopentadienyl ligands. In light of this, if a near-linear dysprosium complex of the type [(Cb)Dy(Cp)] or [Cb2Dy]− can be synthesized, the SMM properties should be competitive with those of current benchmark metallocene systems of the type [Cp2Dy]+.
For financial support, we thank the University of Sussex, the ERC (CoG 646740), the EPSRC (EP/M022064/1), the National Natural Science Foundation of China (Grants 21525103 and 21871247), the Magnus Ehrnrooth Foundation, the CSC-IT Center for Science in Finland, the Finnish Grid and Cloud Infrastructure (urn:nbn:fi:research-infras-2016072533), the University of Oulu (Kvantum Institute) and Prof. H. M. Tuononen (University of Jyväskylä) for computational resources. JT and RAL also thank the Royal Society for a Newton Advanced Fellowship (NA160075).
Conflicts of interest
There are no conflicts to declare.Notes and references
- F.-S. Guo, A. K. Bar and R. A. Layfield, Chem. Rev., 2019, 119, 847 Search PubMed.
- J. Lu, M. Guo and J. Tang, Chem. – Asian J., 2017, 12, 2772 CrossRef CAS PubMed.
- L. Escalera-Moreno, J. J. Baldoví, A. Gaita-Ariño and E. Coronado, Inorg. Chem., 2019, 58, 11883 CrossRef CAS PubMed.
- J. J. Le Roy, J. Cremers, I. A. Thomlinson, M. Slota, W. K. Myers, P. H. Horton, S. J. Coles, H. L. Anderson and L. Bogani, Chem. Sci., 2018, 9, 8474 RSC.
- E. Coronado, Nat. Rev. Mater., 2020, 5, 87 CrossRef.
- J. D. Rinehart and J. R. Long, Chem. Sci., 2011, 2, 2078 RSC.
- F.-S. Guo, B. M. Day, Y.-C. Chen, M.-L. Tong, A. Mansikkamäki and R. A. Layfield, Science, 2018, 362, 1400 CrossRef CAS PubMed.
- B. M. Day, F.-S. Guo and R. A. Layfield, Acc. Chem. Res., 2018, 51, 1880 CrossRef CAS PubMed.
- B. M. Day, F.-S. Guo, S. R. Giblin, A. Sekiguchi, A. Mansikkamäki and R. A. Layfield, Chem. – Eur. J., 2018, 24, 16779 CrossRef CAS PubMed.
- A. Chakraborty, B. M. Day, J. P. Durrant, M. He, J. Tang and R. A. Layfield, Organometallics, 2020, 39, 8 CrossRef CAS.
- N. Tsoureas, A. Mansikkamäki and R. A. Layfield, Chem. Commun., 2020, 56, 944 RSC.
- J. T. Boronski, L. R. Doyle, J. A. Seed, A. J. Wooles and S. T. Liddle, Angew. Chem., Int. Ed., 2020, 59, 295 CrossRef CAS PubMed.
- A. Sekiguchi, T. Matsuo and H. Watanabe, J. Am. Chem. Soc., 2000, 122, 5652 CrossRef CAS.
- B. O. Roos, in Advances in Chemical Physics: Ab Initio Methods in Quantum Chemistry II, ed. K. P. Lawley, Wiley, New York, NY, USA, 1987, vol. 69, pp. 399–455 Search PubMed.
- P. Siegbahn, A. Heiberg, B. Roos and B. Levy, Phys. Scr., 1980, 21, 323 CrossRef CAS.
- B. O. Roos, P. R. Taylor and P. E. M. Siegbahn, Chem. Phys., 1980, 48, 157 CrossRef CAS.
- P. E. M. Siegbahn, J. Almlöf, A. Heiberg and B. O. Roos, J. Chem. Phys., 1981, 74, 2384 CrossRef.
- P. Å. Malmqvist, B. O. Roos and B. Schimmelpfennig, Chem. Phys. Lett., 2002, 357, 230 CrossRef CAS.
- L. F. Chibotaru and L. Ungur, J. Chem. Phys., 2012, 137, 64112 CrossRef CAS PubMed.
- L. Ungur, M. Thewissen, J.-P. Costes, W. Wernsdorfer and L. F. Chibotaru, Inorg. Chem., 2013, 52, 6328 CrossRef CAS PubMed.
- L. Ungur and L. F. Chibotaru, Chem. – Eur. J., 2017, 23, 3708 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Synthesis details, spectroscopic characterization, magnetic measurements and computational details. CCDC 1987236–1987239. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0cc01722a |
| This journal is © The Royal Society of Chemistry 2020 |