
A tetranuclear nickel cluster isolated in multiple high-valent states†
Samuel I.
Jacob
a,
Iskander
Douair
b,
Guang
Wu
a,
Laurent
Maron
 b and
Gabriel
Ménard
*a
b and
Gabriel
Ménard
*a
aDepartment of Chemistry and Biochemistry, University of California, Santa Barbara, CA 93106, USA. E-mail: menard@chem.ucsb.edu
bCNRS, INSA, UPS, UMR 5215, LPCNO, Université de Toulouse, 135 avenue de Rangueil, 31077 Toulouse, France
First published on 15th April 2020
Abstract
We report a series of high-valent tetranuclear nickel clusters isolated from the chemical oxidation of an all Ni(II) ([Ni4]) neutral cluster. Electrochemical analysis of [Ni4] reveals three reversible sequential oxidations at 0.248 V (1e−), 0.678 V (1e−), and 0.991 V (2e−) vs. Fc/Fc+ corresponding to mono-, di-, and tetra-oxidized species, [Ni4]+, [Ni4]2+, [Ni4]4+, respectively. Using spectroscopic, crystallographic, magnetometric, and computational techniques, we assign the primary loci of oxidations to the Ni centers in each case, thus resulting in the isolation of the first tetranuclear all-Ni(III) cluster, [Ni4]4+.
Isolated high-valent nickel complexes (NiIII, NiIV) are relatively rare,1–6 yet are frequently invoked in mechanistic studies involving reductive elimination steps, particularly in C–C and C-heteroatom bond forming chemistry.7–10 Bimetallic complexes with formally high-valent cores, [Ni2]n+ (n > 4), have been shown to engage in hydroxylation chemistry, as well as C-heteroatom and N–N bond forming reactions.11–14 Co-facial, high-valent (Ni(III)) cores ([Ni2]6+) are also proposed to stabilize reactive intermediates through metal–metal bonding interactions,14 similar to more extensively studied heavier [Pd2] congeners.15,16 Co-facial bimetallics have also been extensively studied by Cotton and others17 using the familiar C4-symmetric paddlewheel geometries, including in mixed-valent ([Ni2]5+) and all high-valent ([Ni2]6+) cores, providing significant fundamental insights into Ni–Ni bonding interactions.18,19 While multinuclear Ni complexes ([Nix]; x > 2) are well known20–24 – with some isolated in partial higher-valent states25–28 – to the best of our knowledge, only a few are isolated in all Ni(III) states,29,30 and none are tetranuclear. Tethering multiple high-valent centers together may open the door to mediating multi-electron transformations, and may further shed light on core metal–metal bonding interactions in such expanded motifs. Herein, we describe the synthesis and characterization of a tetranuclear [Ni4]8+ cluster (denoted [Ni4]; Scheme 1), with an orthogonal double paddlewheel core (distorted tetrahedron), as well as its oxidized forms, [Ni4]+, [Ni4]2+, and what we assign as the first all Ni(III) tetranuclear cluster, [Ni4]4+. Spectroscopic, crystallographic, and computational studies support the metal-based Ni(III) states in the tetranuclear core upon sequential oxidations.
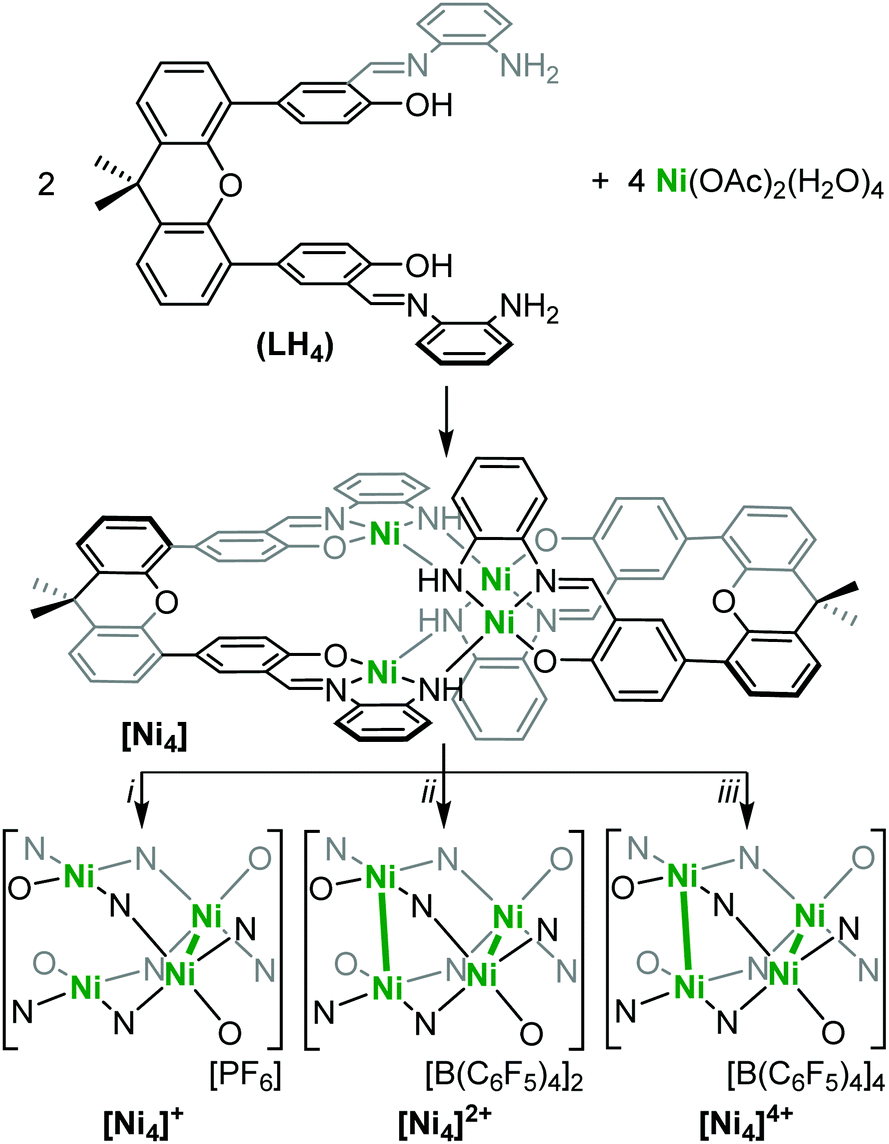 | ||
| Scheme 1 (i) [NO][PF6] (0.9 equiv.), DCM, r.t., 4 h; (ii) [(2,4-Br2C6H3)3N][B(C6F5)4] (2.1 equiv.), DCM, r.t., 15 min; (iii) [(2,4-Br2C6H3)3N][B(C6F5)4] (10 equiv.), DCM, r.t., 10 min. Ni–Ni bonding is indicated by bolded green lines. | ||
In order to target higher nuclearity complexes, we modified a known, dinucleating, xanthene-bridged bis-salen ligand31 by exposing its precursor, 5,5′-(9,9-dimethylxanthene-4,5-diyl)bis(salicylaldehyde), to an excess (10×) of 1,2-diaminobenzene in ethanol under reflux. The product ligand, LH4 (Scheme 1), was cleanly generated in 84% isolated yield following workup and now features two tridentate pockets, making it more amenable to cluster formation. Metalation of LH4 with 2 equiv. of Ni(OAc)2·4H2O afforded the tetranuclear complex, L2Ni4 ([Ni4]), in 50% isolated yield following workup (Scheme 1). The red, diamagnetic, air stable complex was readily characterized by 1H NMR spectroscopy and MALDI mass spectrometry. Red single crystals suitable for X-ray diffraction (XRD) studies were grown by vapor diffusion of hexanes into a concentrated dichloromethane (DCM) solution. The solid-state molecular structure revealed four Ni centers in square planar geometries with the fourth coordination sites filled by amine donating groups from adjacent ligands (Scheme 1 and Fig. 1a, Fig. S15, ESI†). The tetranuclear core features co-facial Ni1–Ni2 and Ni3–Ni4 distances of 2.8662(19) Å and 2.886(2) Å, respectively, and adopts an orthogonal double paddlewheel structure (Fig. 1a and Table 1). The transverse Ni–Ni distances (e.g. Ni1–Ni3) are longer and average 3.33 Å.
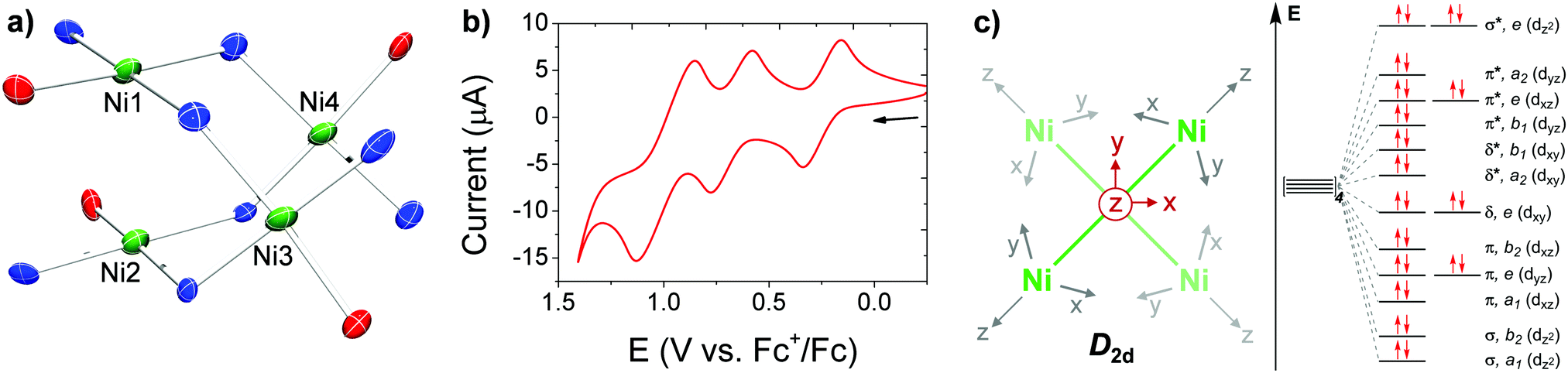 | ||
| Fig. 1 (a) Partial solid-state structure of [Ni4] highlighting the inner core (see Fig. S15 for full structure, ESI†). (b) CV of [Ni4] in TCE (1 mM of [Ni4], 0.1 M [Bu4N][PF6] supporting electrolyte), glassy carbon working electrode, Pt wire counter electrode, and Ag wire pseudo-reference electrode. (c) Partial d-orbital splitting diagram for a D2d tetrametallic core, ignoring ligand π interactions. | ||
| [Ni4] | [Ni4]+ | [Ni4]2+ | [Ni4]4+ | ||
|---|---|---|---|---|---|
| Ni–Ni distance (Å) | Ni1–Ni2 | 2.8662(19) | 2.889(2) | 2.7095(18) | 2.686(3) |
| Ni3–Ni4 | 2.886(2) | 2.640(2) | 2.6284(18) | 2.668(3) | |
| Avg | 2.876 | 2.765 | 2.669 | 2.677 | |
| Ni–mean plane distance (Å) | Ni1 | 0.065 | 0.090 | 0.114 | 0.123 |
| Ni2 | 0.055 | 0.073 | 0.139 | 0.123 | |
| Ni3 | 0.080 | 0.158 | 0.140 | 0.143 | |
| Ni4 | 0.064 | 0.117 | 0.130 | 0.143 | |
| Avg | 0.066 | 0.110 | 0.131 | 0.133 | |
Cyclic voltammetry (CV) of [Ni4] in 1,1,2,2-tetrachloroethane (TCE) revealed three reversible oxidation events at E1/2 values of 0.248, 0.678, and 0.991 V versus the ferrocene/ferrocenium (Fc/Fc+) redox couple (Fig. 1b) in approximate 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1:2 e− events, respectively, as determined by relative integrations of the oxidative curves. Chemical isolation of the mono-oxidized product was performed by treatment of [Ni4] with an equivalent of [NO][PF6] to yield [L2Ni4][PF6] ([Ni4]+; Scheme 1). Single crystals suitable for XRD studies were obtained by layering hexanes on a concentrated DCM solution of [Ni4]+ at −40 °C. The solid-state structure revealed a significantly contracted Ni3–Ni4 distance of 2.640(2) Å, and a relatively unperturbed Ni1–Ni2 distance of 2.889(2) Å, identical within error to the Ni3–Ni4 bond in [Ni4] (Fig. S16, ESI† and Table 1). Corresponding protrusions of the Ni centers relative to their respective mean tridentate ligand planes (excluding bridging donor atom from an adjacent ligand) are consistent with the bond length contraction (Table 1 and Fig. S23, ESI†). The significant shortening of the Ni3–Ni4 distance is consistent with the formation of a long, formal half bond expected from the removal of a single e− from an e-symmetric σ* orbital located along the Ni3–Ni4 vector (Fig. 1c),18,19,32 using a simplified D2d-symmetric tetranuclear model and ignoring extensive ligand π mixing. An overall spin of ½, determined by the Evans method, further confirmed this assignment.33 In addition, analysis of [Ni4]+ by EPR spectroscopy in DCM at 100 K revealed a rhombic spectrum with significant g tensor anisotropy modeled as: gx = 2.01, gy = 2.22, gz = 2.32, and gav = 2.18 (Fig. S12, ESI†), consistent with previously reported Ni-centered oxidation events.3,4,19 While the UV-Vis absorption spectrum of [Ni4]+ is similar to [Ni4], a distinct absorption in the NIR at 1300 nm (ε = 3277 M−1 cm−1) is observed for [Ni4]+ (Fig. S11, ESI†) which we tentatively assign as a charge transfer band (based on its high ε),19,34 likely ligand-to-metal (LMCT) due to the oxidized Ni center. A comproportionation constant (Kc) of ∼2 × 107, obtained from the difference in the first two E1/2 values,35 supports a Robin-Day Class III fully delocalized system, with resulting Ni3(2.5)–Ni4(2.5) oxidation states in [Ni4]+.
:1:2 e− events, respectively, as determined by relative integrations of the oxidative curves. Chemical isolation of the mono-oxidized product was performed by treatment of [Ni4] with an equivalent of [NO][PF6] to yield [L2Ni4][PF6] ([Ni4]+; Scheme 1). Single crystals suitable for XRD studies were obtained by layering hexanes on a concentrated DCM solution of [Ni4]+ at −40 °C. The solid-state structure revealed a significantly contracted Ni3–Ni4 distance of 2.640(2) Å, and a relatively unperturbed Ni1–Ni2 distance of 2.889(2) Å, identical within error to the Ni3–Ni4 bond in [Ni4] (Fig. S16, ESI† and Table 1). Corresponding protrusions of the Ni centers relative to their respective mean tridentate ligand planes (excluding bridging donor atom from an adjacent ligand) are consistent with the bond length contraction (Table 1 and Fig. S23, ESI†). The significant shortening of the Ni3–Ni4 distance is consistent with the formation of a long, formal half bond expected from the removal of a single e− from an e-symmetric σ* orbital located along the Ni3–Ni4 vector (Fig. 1c),18,19,32 using a simplified D2d-symmetric tetranuclear model and ignoring extensive ligand π mixing. An overall spin of ½, determined by the Evans method, further confirmed this assignment.33 In addition, analysis of [Ni4]+ by EPR spectroscopy in DCM at 100 K revealed a rhombic spectrum with significant g tensor anisotropy modeled as: gx = 2.01, gy = 2.22, gz = 2.32, and gav = 2.18 (Fig. S12, ESI†), consistent with previously reported Ni-centered oxidation events.3,4,19 While the UV-Vis absorption spectrum of [Ni4]+ is similar to [Ni4], a distinct absorption in the NIR at 1300 nm (ε = 3277 M−1 cm−1) is observed for [Ni4]+ (Fig. S11, ESI†) which we tentatively assign as a charge transfer band (based on its high ε),19,34 likely ligand-to-metal (LMCT) due to the oxidized Ni center. A comproportionation constant (Kc) of ∼2 × 107, obtained from the difference in the first two E1/2 values,35 supports a Robin-Day Class III fully delocalized system, with resulting Ni3(2.5)–Ni4(2.5) oxidation states in [Ni4]+.
The di-oxidized product was obtained by treating [Ni4] with two equivalents of the ammoniumyl oxidant, [(2,4-Br2C6H3)3N][B(C6F5)4] to yield [L2Ni4][B(C6F5)4]2 ([Ni4]2+; Scheme 1).36 Single crystals suitable for XRD studies were obtained by layering hexanes on a concentrated DCM solution of [Ni4]2+ at −40 °C. The solid-state structure revealed contracted Ni1–Ni2 (2.7095(18) Å) and Ni3–Ni4 (2.6284(18) Å) distances with corresponding protrusions of the Ni centers from the mean tridentate ligand planes (Table 1 and Fig. S17 and S23, ESI†). These observed contractions are consistent with the removal of an electron from each of the e-degenerate σ* orbitals, with each being localized at the separate Ni1–Ni2 and Ni3–Ni4 vectors, forming two Ni(II)–Ni(III) linkages with formal half-bonds in each (Fig. 1c). The resulting expected S = 1 spin state was confirmed by magnetic measurements of [Ni4]2+ performed by SQUID magnetometry (Fig. S13, ESI†). Magnetic susceptibility (χMT) measurements for [Ni4]2+ were collected at variable temperatures (2–300 K) and revealed a plateauing average χMT value of 0.95 in the 50–300 K range, consistent with an S = 1 manifold approximated by the general formula, χMT = [∑Si(Si + 1)]/2. A magnetic exchange value (J) of 36.2 cm−1 was obtained by fitting the data from 2–300 K and confirmed that significant ferromagnetic coupling between Ni centers is present, possibly due to super exchange through the bridging nitrogen atoms. This degree of magnetic exchange in polynuclear high-valent Ni species via super exchange is not unprecedented and even greater magnitudes have been reported.30 The absorption spectrum revealed a red-shifted LMCT absorbance at 1410 nm (ε = 14574 M−1 cm−1) in the NIR, the intensity of which is over four times as intense as the one in [Ni4]+ (Fig. S11, ESI†). Lastly, a Kc value of ∼2 × 105, obtained from the difference in the two highest E1/2 values,35 suggests a more localized electronic structure consistent with a Robin-Day Class II system.
The isolation of the tetra-oxidized species was achieved by treating [Ni4] with 10 equiv. of [(2,4-Br2C6H3)3N][B(C6F5)4] to yield [L2Ni4][B(C6F5)4]4 ([Ni4]4+; Scheme 1). Single crystals suitable for XRD studies were obtained by layering hexanes on a concentrated DCM solution of [Ni4]4+ at −40 °C. Similar to [Ni4]2+, both Ni–Ni distances are contracted relative to [Ni4], and concurrent protrusions of the Ni centers from the mean ligand tridentate planes are observed (Table 1 and Fig. S18, S23, ESI†). However, these effects are less pronounced than in the dicationic [Ni4]2+, and we attribute this distinction to the increased electrostatic repulsion between cationic metal centers in [Ni4]4+ relative to [Ni4]2+.17,37 Attempts to obtain magnetic measurements of [Ni4]4+ were hampered by its extreme instability (vide infra). Nonetheless, the 1H NMR spectrum of in situ-generated [Ni4]4+ revealed a paramagnetic species (Fig. S9, ESI†), likely arising from thermal population of a higher spin state (Fig. 1c). The absorption spectrum of in situ-generated [Ni4]4+ revealed an intense (ε = 22099 M−1 cm−1) red-shifted LMCT band in the NIR at 1468 nm along with a significant shoulder centered at 1662 nm (ε = 20751 M−1 cm−1), significantly more intense than those observed for [Ni4]+ and [Ni4]2+ (Fig. S11, ESI†).
Due to its highly reactive nature, [Ni4]4+ readily decomposes to [Ni4]2+ during and after the reaction workup thereby complicating a thorough characterization. Therefore, we performed DFT calculations in order to shed light on its electronic structure and determine the locus of oxidation. Calculations were first performed on the neutral [Ni4] species at the B3PW91 level of theory to validate the computational method. The optimized structure (see ESI†) is in good agreement with the experimental one with the Ni–Ni distances reproduced to within 0.05 Å for the singlet spin state (all Ni(II)). Wiberg Bond Indexes (WBI) are found to be very small for the Ni–Ni interactions (0.08). In order to get insight into the locus of oxidation of [Ni4], the frontier orbitals were scrutinized. As can be seen (Fig. S25, ESI†), the HOMO (364) and HOMO−1(363) are mainly antibonding (σ*) Ni–Ni interactions (Fig. 1c), whereas the HOMO−2 (362) is mainly located on the ligand. Therefore, one would expect the first four oxidations to remove electrons from the HOMO and HOMO−1 which mainly occur at the Ni centers. Removing electrons from these two orbitals would lead to a decrease of the Ni–Ni antibonding interaction and a concurrent decrease in the Ni–Ni bond distance, as observed experimentally. In order to verify this assumption, calculations were performed on [Ni4]4+. The optimized structure has a quintet spin state – confirming the observed paramagnetic nature of [Ni4]4+ (vide supra) – and is in good agreement with the experimental structure with the Ni–Ni distances reproduced to within 0.02 Å, roughly 0.2 Å shorter than in [Ni4]. This is further highlighted by the increase of the WBI of the Ni–Ni interactions (0.2) indicating a stronger interaction. In order to assess the oxidation state of the Ni centers, the unpaired spin density was plotted (Fig. 2). As can be seen, the unpaired spin density is primarily located on the Ni centers, with some contribution from the oxygen on the ligand, thus supporting our assignment as an all high-valent (all Ni(III)) multinuclear cluster in [Ni4]4+.
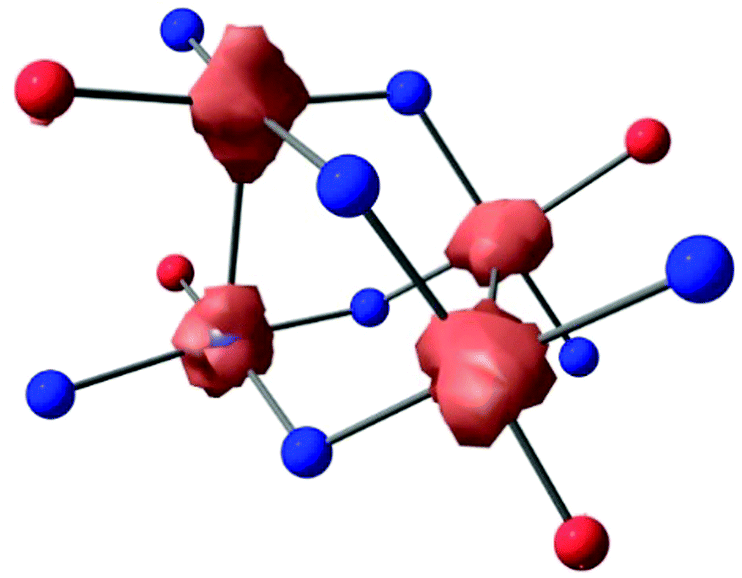 | ||
| Fig. 2 Unpaired spin density plot for [Ni4]4+. The isocontour value is set to the default (0.03). | ||
We would lastly like to acknowledge that o-phenylenediamine and Schiff base (i.e. salen) derivatives are known redox-active ligands and experience distinct site-specific alterations in bond lengths at either Ni–N/O bonds, or within the ligand multiple bonds upon oxidation.38–41 A thorough analysis of the bond lengths in the neutral ([Ni4]), as well as oxidized complexes ([Ni4]n+; n = 1, 2, 4), revealed a general shortening of the Ni–N/O bonds upon oxidation – expected from metal-based oxidations3,19 – but no discernable bond change patterns within the ligand π framework expected from ligand-based oxidation (Table S1 and Fig. S19–S22, ESI†). With this, and together with our combined experimental and theoretical results above, we propose that [Ni4] undergoes primarily metal-based oxidation events.
In conclusion, a novel tetranuclear Ni species isolated in multiple high-valent states, including an all Ni(III) state, is reported and supported by spectroscopic, crystallographic, magnetometric, and computational data. We are currently investigating potential applications of this cluster in mediating multi-electron transformations at small molecules of energy importance.
CalMip is gratefully acknowledged for a generous grant of computing time. L. M. is a senior member of the Institut Universitaire de France, and acknowledges the Humboldt foundation and Chinese Academy of Science. The University of California, Santa Barbara, as well as the Hellman Fellows Fund are thanked for financial support.
Conflicts of interest
There are no conflicts to declare.Notes and references
- T. J. Collins, T. R. Nichols and E. S. Uffelman, J. Am. Chem. Soc., 1991, 113, 4708–4709 CrossRef CAS.
- P. J. Alonso, A. B. Arauzo, M. A. García-Monforte, A. Martín, B. Menjón, C. Rillo and M. Tomás, Chem. – Eur. J., 2009, 15, 11020–11030 CrossRef CAS PubMed.
- T.-P.-A. Cao, G. Nocton, L. Ricard, X. F. Le Goff and A. Auffrant, Angew. Chem., Int. Ed., 2013, 53, 1368–1372 CrossRef PubMed.
- M. Eckshtain-Levi, M. Orio, R. Lavi and L. Benisvy, Dalton Trans., 2013, 42, 13323–13326 RSC.
- M. Kouno, N. Yoshinari, N. Kuwamura, K. Yamagami, A. Sekiyama, M. Okumura and T. Konno, Angew. Chem., Int. Ed., 2017, 56, 13762–13766 CrossRef CAS PubMed.
- V. M. Iluc, A. J. M. Miller, J. S. Anderson, M. J. Monreal, M. P. Mehn and G. L. Hillhouse, J. Am. Chem. Soc., 2011, 133, 13055–13063 CrossRef CAS PubMed.
- B. M. Rosen, K. W. Quasdorf, D. A. Wilson, N. Zhang, A.-M. Resmerita, N. K. Garg and V. Percec, Chem. Rev., 2010, 111, 1346–1416 CrossRef PubMed.
- S. Z. Tasker, E. A. Standley and T. F. Jamison, Nature, 2014, 509, 299–309 CrossRef CAS PubMed.
- N. M. Camasso and M. S. Sanford, Science, 2015, 347, 1218–1220 CrossRef CAS PubMed.
- W. Zhou, J. W. Schultz, N. P. Rath and L. M. Mirica, J. Am. Chem. Soc., 2015, 137, 7604–7607 CrossRef CAS PubMed.
- K. Shiren, S. Ogo, S. Fujinami, H. Hayashi, M. Suzuki, A. Uehara, Y. Watanabe and Y. Moro-oka, J. Am. Chem. Soc., 2000, 122, 254–262 CrossRef CAS.
- K. Honda, J. Cho, T. Matsumoto, J. Roh, H. Furutachi, T. Tosha, M. Kubo, S. Fujinami, T. Ogura, T. Kitagawa and M. Suzuki, Angew. Chem., Int. Ed., 2009, 48, 3304–3307 CrossRef CAS PubMed.
- J. B. Diccianni, C. Hu and T. Diao, Angew. Chem., Int. Ed., 2016, 55, 7534–7538 CrossRef CAS PubMed.
- J. B. Diccianni, C. Hu and T. Diao, Angew. Chem., Int. Ed., 2017, 56, 3635–3639 CrossRef CAS PubMed.
- D. C. Powers and T. Ritter, Acc. Chem. Res., 2012, 45, 840–850 CrossRef CAS PubMed.
- D. C. Powers and T. Ritter, Nat. Chem., 2009, 1, 302–309 CrossRef CAS PubMed.
- Multiple Bonds Between Metal Atoms, ed. F. A. Cotton, C. A. Murillo and R. A. Walton, Springer Science and Business Media Inc., New York, NY, 2005 Search PubMed.
- F. A. Cotton, M. Matusz, R. Poli and X. Feng, J. Am. Chem. Soc., 1988, 110, 1144–1154 CrossRef CAS.
- J. F. Berry, E. Bothe, F. A. Cotton, S. A. Ibragimov, C. A. Murillo, D. Villagrán and X. Wang, Inorg. Chem., 2006, 45, 4396–4406 CrossRef CAS PubMed.
- J. L. Davidson, M. Green, F. G. A. Stone and A. J. Welch, J. Am. Chem. Soc., 1975, 97, 7490–7492 CrossRef CAS.
- C. G. Efthymiou, C. P. Raptopoulou, A. Terzis, R. Boča, M. Korabic, J. Mrozinski, S. P. Perlepes and E. G. Bakalbassis, Eur. J. Inorg. Chem., 2006, 2236–2252 CrossRef CAS.
- M. Fondo, N. Ocampo, A. M. García-Deibe, R. Vicente, M. Corbella, M. R. Bermejo and J. Sanmartín, Inorg. Chem., 2006, 45, 255–262 CrossRef CAS PubMed.
- F. A. Cotton, C. A. Murillo and Q. Wang, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2007, 63, m1905 CrossRef CAS.
- M. A. Lemes, G. Brunet, A. Pialat, L. Ungur, I. Korobkov and M. Murugesu, Chem. Commun., 2017, 53, 8660–8663 RSC.
- J. F. Berry, F. A. Cotton, L. M. Daniels and C. A. Murillo, J. Am. Chem. Soc., 2002, 124, 3212–3213 CrossRef CAS PubMed.
- T. E. North, J. B. Thoden, B. Spencer and L. F. Dahl, Organometallics, 1993, 12, 1299–1313 CrossRef CAS.
- G.-C. Huang, S.-A. Hua, I. P.-C. Liu, C.-H. Chien, J.-H. Kuo, G.-H. Lee and S.-M. Peng, C. R. Chim., 2012, 15, 159–162 CrossRef CAS.
- J. F. Berry, F. A. Cotton, P. Lei, T. Lu and C. A. Murillo, Inorg. Chem., 2003, 42, 3534–3539 CrossRef CAS PubMed.
- C.-M. Lee, T.-W. Chiou, H.-H. Chen, C.-Y. Chiang, T.-S. Kuo and W.-F. Liaw, Inorg. Chem., 2007, 46, 8913–8923 CrossRef CAS PubMed.
- T. Beissel, F. Birkelbach, E. Bill, T. Glaser, F. Kesting, C. Krebs, T. Weyhermüller, K. Wieghardt, C. Butzlaff and A. X. Trautwein, J. Am. Chem. Soc., 1996, 118, 12376–12390 CrossRef CAS.
- M. Hirotsu, N. Ohno, T. Nakajima, C. Kushibe, K. Ueno and I. Kinoshita, Dalton Trans., 2010, 39, 139–148 RSC.
- J. F. Berry, Acc. Chem. Res., 2016, 49, 27–34 CrossRef CAS PubMed.
- D. F. Evans, J. Chem. Soc., 1959, 2003–2005 RSC.
- I. Mustieles Marín, T. Cheisson, R. Singh-Chauhan, C. Herrero, M. Cordier, C. Clavaguéra, G. Nocton and A. Auffrant, Chem. – Eur. J., 2017, 23, 17940–17953 CrossRef PubMed.
- P. Zanello, Inorganic Electrochemistry: Theory, Practice and Application, The Royal Society of Chemistry, Cambridge, UK, 2003 Search PubMed.
- K. Lam and W. E. Geiger, J. Org. Chem., 2013, 78, 8020–8027 CrossRef CAS PubMed.
- F. A. Cotton, L. M. Daniels, X. Feng, D. J. Maloney, J. H. Matonic and C. A. Murillio, Inorg. Chim. Acta, 1997, 256, 291–301 CrossRef CAS.
- P. Chaudhuri, C. N. Verani, E. Bill, E. Bothe, T. Weyhermüller and K. Wieghardt, J. Am. Chem. Soc., 2001, 123, 2213–2223 CrossRef CAS PubMed.
- E. Bill, E. Bothe, P. Chaudhuri, K. Chlopek, D. Herebian, S. Kokatam, K. Ray, T. Weyhermüller, F. Neese and K. Wieghardt, Chem. – Eur. J., 2005, 11, 204–224 CrossRef PubMed.
- T. Storr, E. C. Wasinger, R. C. Pratt and T. D. P. Stack, Angew. Chem., Int. Ed., 2007, 46, 5198–5201 CrossRef CAS PubMed.
- R. M. Clarke, T. Jeen, S. Rigo, J. R. Thompson, L. G. Kaake, F. Thomas and T. Storr, Chem. Sci., 2018, 9, 1610–1620 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Synthetic procedures, spectroscopic data, magnetic data, crystallographic data, DFT. CCDC 1978084–1978087. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0cc01699c |
| This journal is © The Royal Society of Chemistry 2020 |