
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Copper catalysed oxidative α-sulfonylation of branched aldehydes using the acid enhanced reactivity of manganese(IV) oxide†
Joe I.
Higham
 and
James A.
Bull
*
and
James A.
Bull
*
Department of Chemistry, Imperial College London, Molecular Sciences Research Hub, White City Campus, Wood Lane, London W12 0BZ, UK. E-mail: j.bull@imperial.ac.uk
First published on 17th March 2020
Abstract
The oxidative coupling of secondary aldehydes and sulfinate salts is achieved using copper catalysis to form α-sulfonyl aldehydes. The use of an acidic co-solvent is important to adjust the oxidation potential of MnO2 as an oxidant. A broad range of sulfonylated aldehydes is prepared, and their further functionalisation is demonstrated. A dual ionic/radical pathway mechanism is proposed.
Oxidative cross-coupling reactions are formally a coupling between two nucleophiles (Fig. 1a), with the Chan–Lam coupling as a notable copper-mediated example.1 The choice of oxidant is crucial to effectively remove excess electrons from the system. In the case where either, or both, coupling partners and the product could be oxidised in a non-productive manner, requirements are narrow to avoid side reactions. Sulfinate salts are attractive nucleophiles to use in oxidative couplings as readily available, easily handled solids, for the direct preparation of medicinally important sulfones,2 but can themselves be oxidised in an unproductive manner. α-Quaternary sulfones feature in several medicinally relevant compounds (Fig. 1b).3–6 Examples include CB2 agonists,3 5-lipoxygenase-activating protein (FLAP) modulators,4 RIP2 kinase inhibitors,5 and matrix metalloprotein inhibitors which have shown efficacy in the treatment of cancer, arthritis and cardiovascular disease.6 There are few direct methods used for their synthesis and current routes require multiple steps: thioalkylation then oxidation, double alkylation of a sulfonyl ester/nitrile, reaction of a pre-formed enolate with a sulfonyl fluoride under cryogenic conditions,3–6 rearrangement of vinyl tosylates,7 or rearrangement of an epoxysulfone.8 The oxidative coupling of sulfinate salts to form α- and β-sulfonylketones has been demonstrated using aryl-ethyl-ketones,9 and tertiary cyclopropanols respectively.10,11 The oxidative cross-coupling of aldehydes and sulfinate salts forming α-sulfonyl aldehydes directly is not known. The coupling of branched aldehydes with sulfinate salts would allow rapid access to α-sulfonyl aldehydes in a single step and provide a valuable handle to install directly the type of adjacent functionality displayed in the compounds in Fig. 1.
 | ||
| Fig. 1 (a) Schematic oxidative coupling. (b) Medicinally relevant quaternary sulfones. (c) This work detailing a method to access quaternary sulfonyl–aldehydes via a key oxidative coupling. | ||
Notably the targeted coupling involves three potentially oxidatively sensitive species, presenting an interesting challenge to identify an oxidant of high selectivity to achieve product formation without degradation. Here we report a realisation of this approach using MnO2 in the presence of acetic acid to enhance the redox potential.
Initially, we examined copper catalysed processes along with a range of oxidants, intending to form Cu enolates to intercept with sulfonyl radicals. An initial hit was achieved using silver acetate as an oxidant, and the reaction conditions were optimised examining all parameters (see ESI,† for full discussion). Effective conditions for the coupling of cyclohexane carboxaldehyde 1 with p-tolylSO2Na 2a were achieved using the combination of copper triflate with bathophenanthroline ligand, and silver acetate (Scheme 1a). A solvent mixture of water and ethanol was most productive, ensuring homogeneity of the reaction mixture. This reaction was optimised to provide 69% of the coupled product, however, other aryl sulfinate salts were significantly less reactive (Scheme 1b), and the yields were not improved by modifying reaction conditions or increasing time. There was an apparent correlation between yield and the electrode potential of the sulfinate salt (Scheme 1c).12 In order to improve the scope of the reaction, a stronger oxidant was needed, preferably one which could be fine-tuned to maximise the efficiency of the coupling.
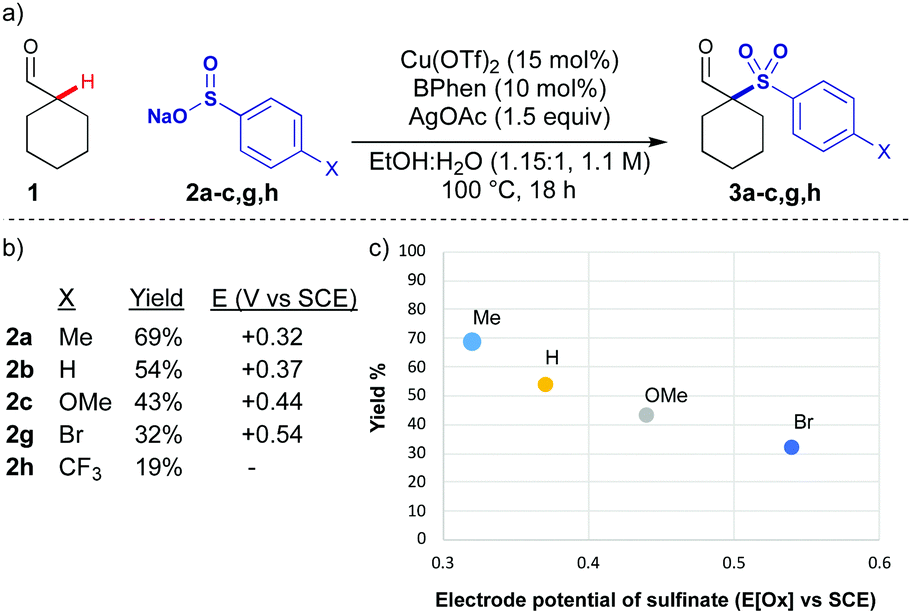 | ||
| Scheme 1 (a) Silver mediated oxidative sulfonylation of cyclohexane carboxaldehyde 1. (b) Yield of sulfone and electrode potential of sulfinate salt.12 (c) Correlation of yield and electrode potential with silver mediated conditions. Reactions performed on 0.4 mmol scale. All yields are isolated yields. | ||
Strong oxidants, such as potassium persulfate, led to over-oxidation. Instead, we chose to examine MnO2, which gave low yields initially but was compatible with the reaction conditions. The oxidative ability of MnO2 is known to change with pH, which can be understood by its redox couple (eqn (1)).
| [MnO2 + 4H+ + 2e− → Mn2+ + 2H2O] | (1) |
A broad range of electrode potentials can be accessed by tuning the pH of the system [pH 4–0, 0.8–1.2 V].13 This property has been little explored in synthetic studies, therefore MnO2 was selected for further investigation.
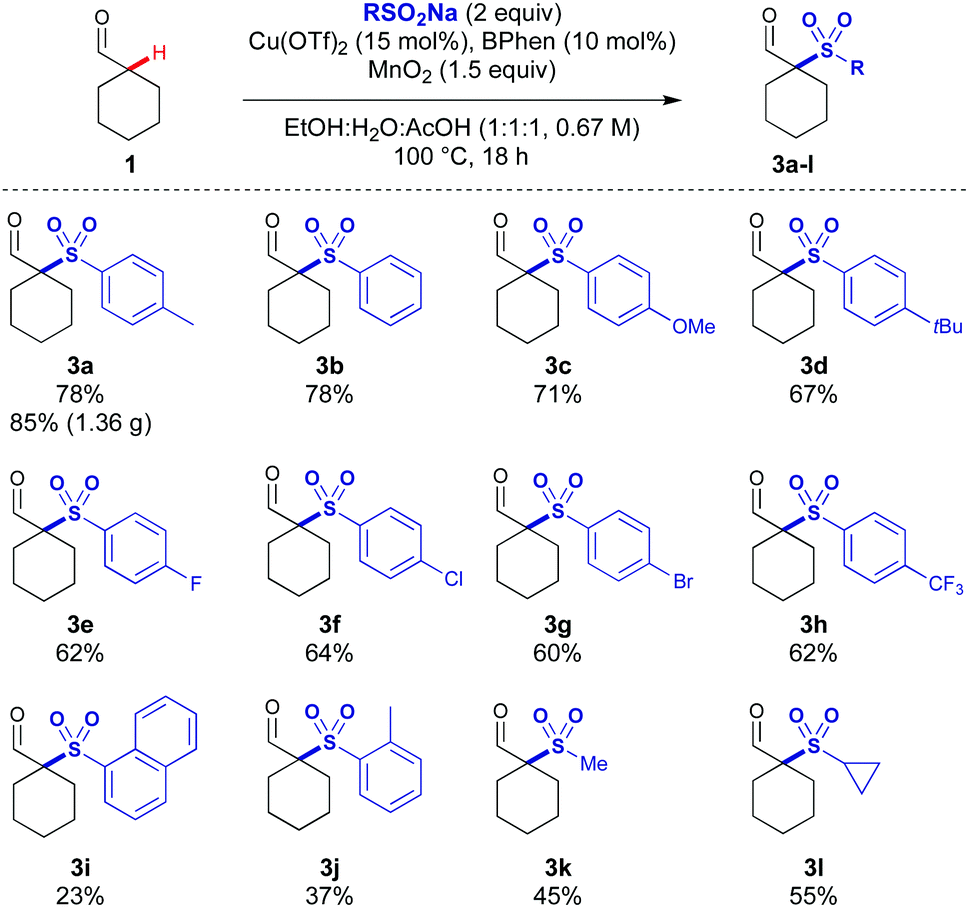
Several acidic additives were screened with a range of sulfinates, and acetic acid was identified as an effective additive. Using acetic acid as a co-solvent gave 71% of the sulfonylated adduct 3a using cyclohexylcarboxaldehyde 1 and p-tolylSO2Na 2a (Table 1, entry 1). In the absence of acetic acid, a low yield was observed (entry 2). In the absence of MnO2 the reaction proceeds in low yield, indicating oxygen from air can facilitate the reaction and some turnover of the Cu catalyst (entry 3). A lower loading of AcOH led to a small decrease in yield (entry 4). When using trichloroacetic acid, no product formation was observed, potentially due to the lower pH leading to the MnO2 being rendered too oxidising leading to product and substrate degradation (entry 5). Under ligandless conditions, a significant reduction in yield was observed emphasising its importance in the reaction (entry 6). The reaction was still observed in the absence of copper, indicating that under MnO2 conditions there is a copper-independent mechanism for product formation (entry 7).14 A slight decrease in reaction yield was observed when the reaction was run under argon indicating that while air is beneficial it has only a minor role in reactivity (entry 8). Unexpectedly, the reaction proceeded in the presence of radical traps (TEMPO and dihydroanthracene) without significant loss in yield (entries 9 and 10). This suggests that the reaction could be proceeding by multiple reaction pathways. When increasing the scale of the reaction, a further increase in yield was observed, potentially due to reduced losses from evaporation, affording 85% of sulfonyl-aldehyde 3a on 6 mmol scale (Scheme 2).
|
|
||
|---|---|---|
| Entry | Change to standard conditions | Yield 3aa (%) |
| Reactions performed on 0.2 mmol scale.a Yield determined by 1H NMR using 1,3,5-trimethoxybenzene as an internal standard. | ||
| 1 | None | 71 |
| 2 | No AcOH | 27 |
| 3 | No MnO2 | 29 |
| 4 | 4 equiv. AcOH | 63 |
| 5 | Cl3CCO2H (10 equiv.) instead of AcOH | 0 |
| 6 | No BPhen | 35 |
| 7 | No Cu | 21 |
| 8 | Under Ar | 61 |
| 9 | +2 equiv. TEMPO | 50 |
| 10 | +2 equiv. dihydroanthracene | 42 |
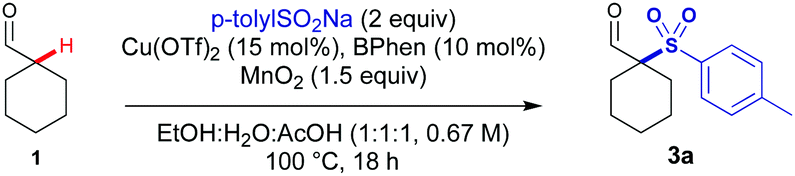
 | ||
| Scheme 2 Reaction scope varying the sulfinate salts. Reactions performed on 0.4 mmol scale. All yields are of the isolated product. | ||
With these conditions, we next investigated the tolerance of the reaction to substrate changes (Scheme 2). Improved yields were observed for all aryl sulfinate salts compared to the silver mediated conditions. Electron-neutral (phenyl) and aryl groups with electron-rich substituents (p-OMe and p-tBu) gave 3b–3d in excellent yields. Aryl sulfinates with p-F, p-Cl and p-Br substituents reacted effectively under these conditions (3e–3g), with a significant increase in the yield of 3g (60% vs. 32%, Mn vs. Ag oxidants).
Importantly, the most challenging electron poor p-trifluoromethylphenyl sulfinate salt reacted effectively under these conditions to yield 3h in 62% (a threefold increase). More sterically demanding sulfinate salts (1-naphthyl and o-tolyl) are also reactive affording 3i and 3j, albeit in lower yields. Methyl and cyclopropane sulfinate salts could be used to access dialkyl sulfones 3k and 3l.
The scope of the aldehydes proved to be broad (Scheme 3). Ring sizes from 4 to 8 could be coupled to give the quaternary sulfonylated products 4a–7a, with higher yields observed for the less strained ring systems. 4-tert-Butylcyclohexane carboxaldehyde was sulfonylated in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture of cis- and trans-diastereoisomers to give 8a, which is consistent with the involvement of a planar intermediate derived from the aldehyde in the reaction mechanism. Aldehydes bearing phenyl, CH2OBn and ester substituents were also successful (9a–11a). Heterocyclic THP and Cbz-protected piperidine carboxaldehydes were compatible to give 12a and 13a which are attractive building blocks for use in medicinal chemistry.
:1 mixture of cis- and trans-diastereoisomers to give 8a, which is consistent with the involvement of a planar intermediate derived from the aldehyde in the reaction mechanism. Aldehydes bearing phenyl, CH2OBn and ester substituents were also successful (9a–11a). Heterocyclic THP and Cbz-protected piperidine carboxaldehydes were compatible to give 12a and 13a which are attractive building blocks for use in medicinal chemistry.
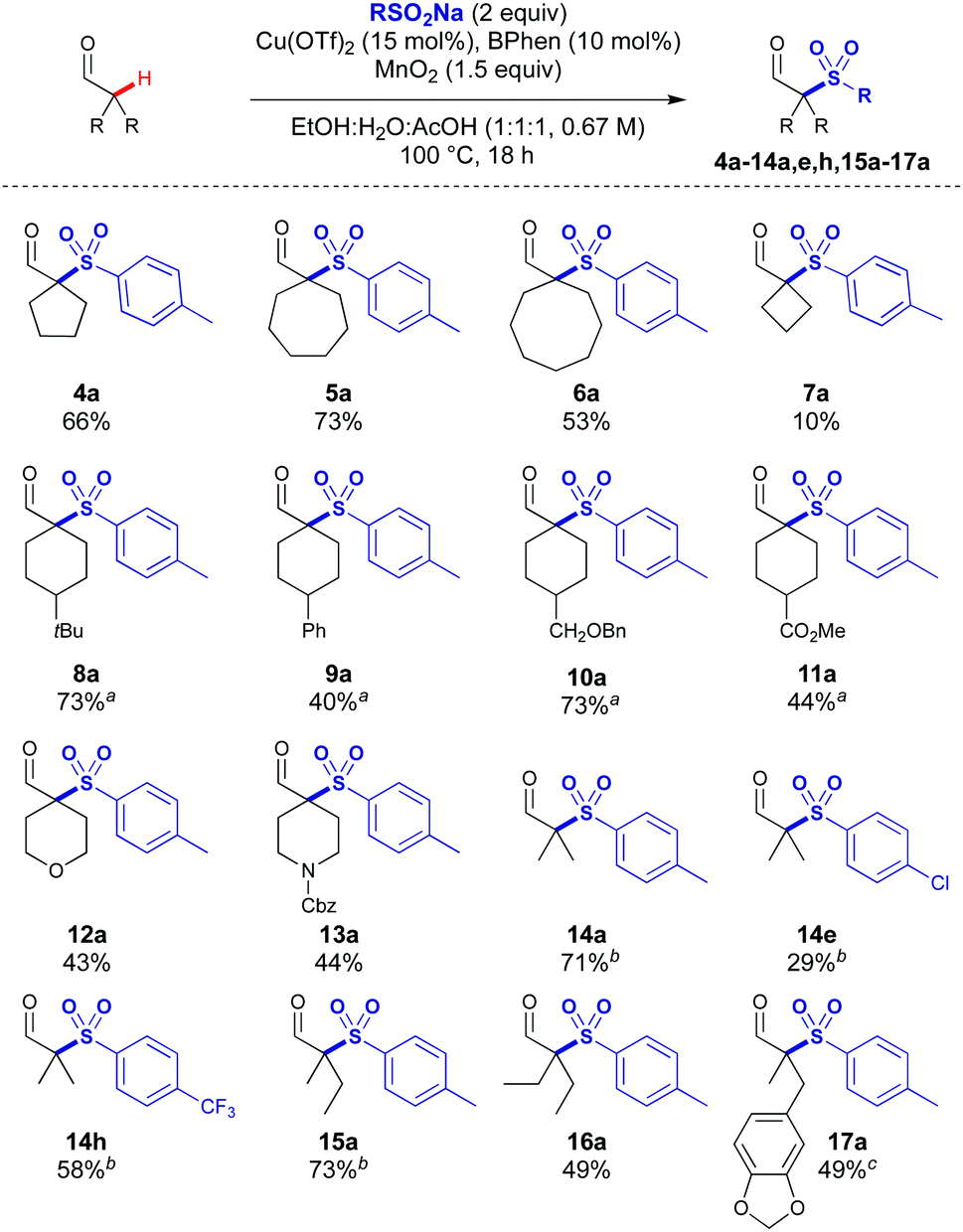 | ||
| Scheme 3 Reaction scope varying the aldehyde. All reactions performed on 0.4 mmol scale. All yields are of the isolated product. a1:1 dr determined by 1H NMR. bReaction performed on 1 mmol scale. cBasic aqueous workup performed to avoid product instability. | ||
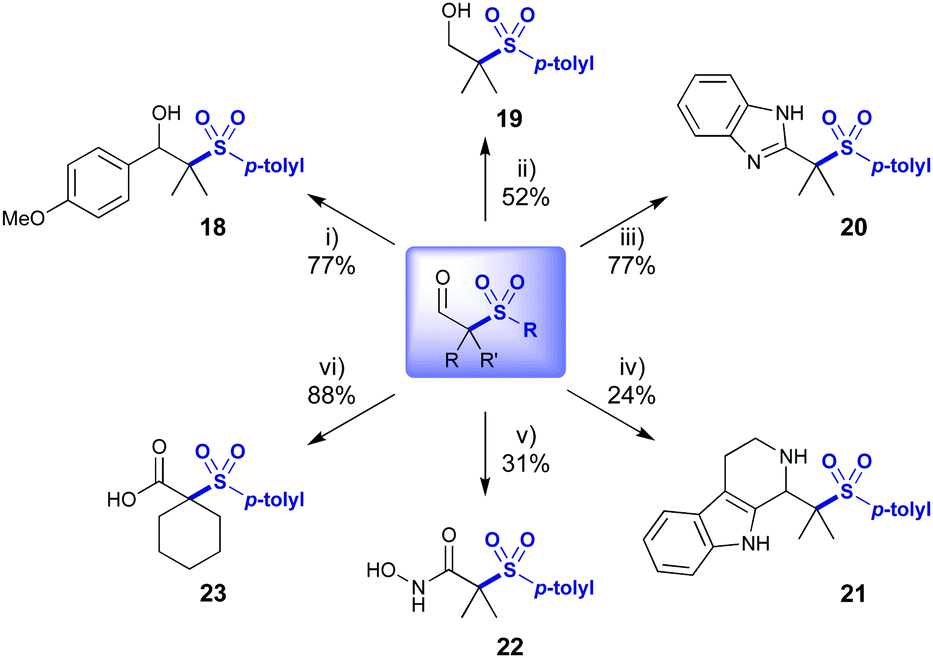 | ||
| Scheme 4 Derivatisation of sulfonated aldehyde. (i) p-MeOC6H4MgBr, THF, 0 °C. (ii) NaBH4, EtOH, 0 °C. (iii) o-Phenylenediamine, CAN (10 mol%), 30% H2O2 in H2O (4 equiv.), MeCN, 50 °C. (iv) Tryptamine, TFA, CH2Cl2, r.t. (v) N-Hydroxysuccinimide and PhI(OAc)2, 0 °C, MeCN; then NH2OH·HCl (2 equiv., 50 w/w% in H2O), r.t. (vi) NaClO2 (4 equiv.), NaH2PO4 (2.2 equiv.), H2O2 (1 equiv.) in MeCN/H2O, r.t. | ||
Acyclic substrates were effective which included gem-dimethyl products 14a, 14e and 14h bearing p-Me, p-Cl and p-CF3 substituents, as the gem-dimethyl motif is a common substructure in bioactive compounds. Increasing the chain length of the aldehyde was tolerated giving 15a and 16a in 73% and 49% respectively. Benzodioxazole substituted aldehyde 17a could be accessed without any observed hydrolysis of the acetal or oxidation of the benzylic position.
Exploitation of the aldehyde functionality enabled divergent access to more complex sulfone derivatives in a single step (Scheme 4). Addition of a Grignard reagent afforded benzylic alcohol 18 in 77% yield. Reduction with NaBH4 afforded alcohol 19 and oxidative cyclisation with o-phenylene diamine afforded benzimidazole 20 in excellent yield.15 A Pictet–Spengler reaction gave access to indole containing 21.16 Hydroxamic acid 22 was synthesised by oxidative amination of the aldehyde.17 Aldehyde 3a was oxidised with a Pinnick oxidation to acid 23, itself providing a valuable building block.18
Based on the above results and prior literature, we propose a plausible dual mechanistic pathway involving separate ionic and radical steps (Scheme 5).11c,19 Both pathways involve coordination of the aldehyde to CuII complex A to form cationic complex B. Coordination of the aldehyde acidifies the α-position, allowing deprotonation of B to form the copper enol/enolate C. At this point, the mechanisms diverge: in the ionic mechanism, the copper centre could be oxidised to CuIII intermediate D which could then be attacked by the sulfinate to give the product aldehyde and CuI intermediate E. Alternatively the product could be formed by direct attack of intermediate C by a sulfonyl radical formed by oxidation of the sulfinate salt.
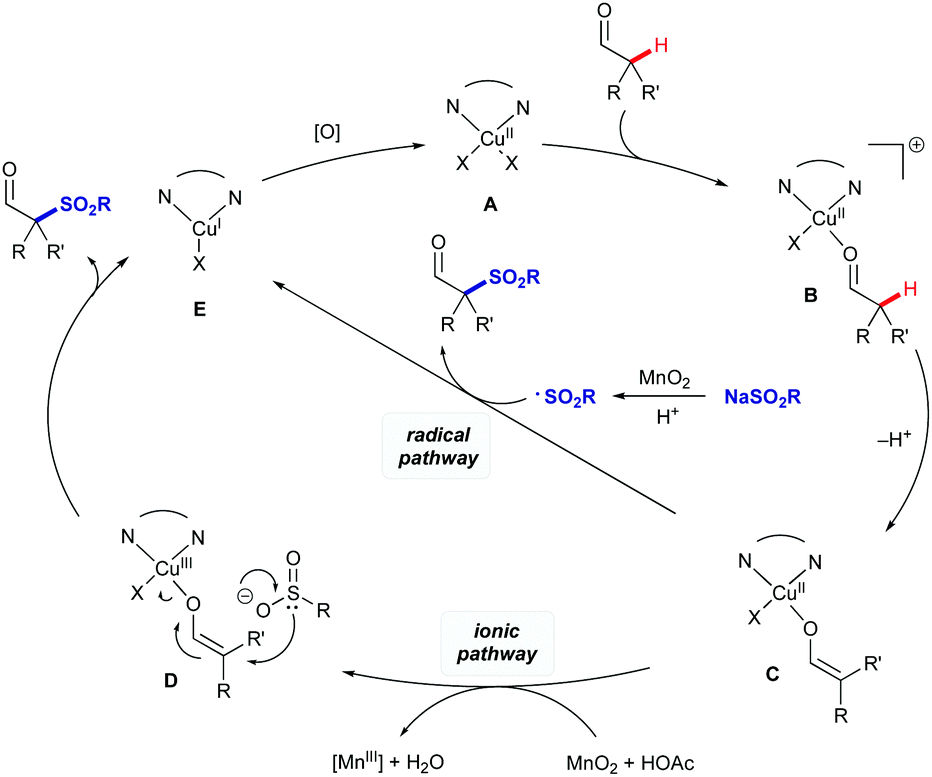 | ||
| Scheme 5 Proposed mechanism of α-sulfonylation. | ||
In summary, a new α-sulfonylation reaction of secondary aldehydes has been achieved using earth abundant copper catalysis. Manganese(IV) oxide was an effective oxidant when using AcOH to enhance the redox potential. These conditions were compatible with the potentially sensitive aldehyde products and a wide range of sulfinate salts. Control and radical trapping experiments indicate this process could proceed by concurrent polar and radical mechanisms. The utility of these aldehyde building blocks was shown by their derivatisation into functionally diverse fragments, suggesting this methodology may of value in medicinal chemistry programs.
We gratefully acknowledge The Royal Society for funding [University Research Fellowship, UF140161 (to J. A. B.), URF appointed grant RG150444 and URF enhancement grant RGF\EA\180031].
Conflicts of interest
There are no conflicts to declare.Notes and references
- For reviews on oxidative couplings, see: (a) I. Funes-Ardoiz and F. Maseras, ACS Catal., 2018, 8, 1161–1172 CrossRef CAS; (b) W. Shi, C. Liu and A. Lei, Chem. Soc. Rev., 2011, 40, 2761–2776 RSC; (c) For a review of Chan–Lam coupling see: M. J. West, J. W. B. Fyfe, J. C. Vantourout and A. J. B. Watson, Chem. Rev., 2019, 119, 12491–12523 CrossRef CAS PubMed.
- J. Aziz, S. Messaoudi, M. Alami and A. Hamze, Org. Biomol. Chem., 2014, 12, 9743–9759 RSC.
- E. R. Hickey, D. Riether, D. S. Thomson, R. M. Zindell, P. Amouzegh, M. Ermann, C. F. Palmer and M. Whittaker, Sulfone Compounds Which Modulate the CB2 Receptor, US Pat. US20120015988A1, 2010 Search PubMed.
- G. Sales, M. Bacani, W. Eccles, A. E. Fitzgerald, S. D. Goldberg, M. D. Hack, N. A. Hawryluk, W. M. Jones, J. M. Keith, P. Krawczuk, A. D. Lebsack, A. Lee-Dutra, J. Liu, K. J. McClure, S. P. Meduna, D. J. Pippel, M. D. Rosen and Z. S. Sales, Flap Modulators, US Pat. US9079866B2, 2015 Search PubMed.
- M. J. Bury, L. N. Casillas, A. K. Charnley, P. A. Haile, W. Marquis Jr, J. F. Mehlmann, J. J. Romano, R. R. Singhous Jr and G. Z. Wang, Amino-quinolines as kinase inhibitors, WO 2012/122011 A2, 2012 Search PubMed.
- (a) D. P. Becker, T. E. Barta, L. J. Bedell, T. L. Boehm, B. R. Bond, J. Carroll, C. P. Carron, G. A. Decrescenzo, A. M. Easton, J. N. Freskos, C. L. Funckes-Shippy, M. Heron, S. Hockerman, C. P. Howard, J. R. Kiefer, M. H. Li, K. J. Mathis, J. J. McDonald, P. P. Mehta, G. E. Munie, T. Sunyer, C. A. Swearingen, C. I. Villamil, D. Welsch, J. M. Williams, Y. Yu and J. Yao, J. Med. Chem., 2010, 53, 6653–6680 CrossRef CAS PubMed; (b) J. Orbe, J. A. Sánchez-Arias, O. Rabal, J. A. Rodríguez, A. Salicio, A. Ugarte, M. Belzunce, M. Xu, W. Wu, H. Tan, H. Ma, J. A. Páramo and J. Oyarzabal, J. Med. Chem., 2015, 58, 2465–2488 CrossRef CAS PubMed; (c) I. D. Miguel, J. Orbe, J. A. Sánchez-Arias, J. A. Rodríguez, A. Salicio, O. Rabal, M. Belzunce, E. Sáez, M. Xu, W. Wu, H. Tan, H. Ma, J. A. Páramo and J. Oyarzabal, ACS Med. Chem. Lett., 2018, 9, 428–433 CrossRef PubMed.
- (a) L. Yi, L. Mai and Y. Dongwei, Method for efficiently synthesizing beta-ketone sulfonyl compounds through visible light induction, CN106905099, 2017 Search PubMed; (b) L. Xie, X. Zhen, S. Huang, X. Su, M. Lin and Y. Li, Green Chem., 2017, 19, 3530–3534 RSC.
- T. Durst and K. C. Tin, Tetrahedron Lett., 1970, 11, 2369–2372 CrossRef.
- X.-W. Lan, N.-X. Wang, C.-B. Bai, W. Zhang, Y. Xing, J.-L. Wen, Y.-J. Wang and Y.-H. Li, Sci. Rep., 2015, 5, 18391 CrossRef PubMed.
- Y. A. Konik, G. Z. Elek, S. Kaabel, I. Järving, M. Lopp and D. G. Kananovich, Org. Biomol. Chem., 2017, 15, 8334–8340 RSC.
- (a) X. Tang, L. Huang, Y. Xu, J. Yang, W. Wu and H. Jiang, Angew. Chem., Int. Ed., 2014, 53, 4205–4208 CrossRef CAS PubMed; (b) For sulfonamide synthesis with sulfinate salts, see: X. Tang, L. Huang, C. Qi, X. Wu, W. Wu and H. Jiang, Chem. Commun., 2013, 49, 6102–6104 RSC; (c) For an oxidative alkylation of aldehydes using copper catalysis, see: H. Richter, R. Rohlmann and O. G. Mancheño, Chem. – Eur. J., 2011, 17, 11622–11627 CrossRef CAS PubMed.
- For electrode potentials of sulfinate salts, see: A. U. Meyer, K. Straková, T. Slanina and B. König, Chem. – Eur. J., 2016, 22, 8694–8699 CrossRef CAS PubMed.
- For pourbaix diagram of Manganese see: (a) O. V. Boytsova, T. O. Shekunova and A. E. Baranchikov, Russ. J. Inorg. Chem., 2015, 60, 546–551 CrossRef CAS; (b) J. Hem, US Geol. Surv. Water-Supply Pap., 1963, 1667-A Search PubMed.
- Only trace product was observed under silver mediated conditions in the absence of copper.
- Procedure adapted from K. Bahrami, M. M. Khodaei and F. Naali, J. Org. Chem., 2008, 73, 6835–6837 CrossRef CAS PubMed.
- Procedure for Pictet–Spengler reaction using tertiary aldehydes adapted from: Z. Zhao, Y. Sun, L. Wang, X. Chen, Y. Sun, L. Lin, Y. Tang, F. Li and D. Chen, Tetrahedron Lett., 2019, 60, 800–804 CrossRef CAS.
- G. Dettori, S. Gaspa, A. Porcheddu and L. D. Luca, Adv. Synth. Catal., 2014, 356, 2709–2713 CrossRef CAS.
- Procedure using H2O2 as HOCl scavenger for Pinnick oxidation adapted from: E. Dalcanale and F. Montanari, J. Org. Chem., 1986, 51, 567–569 CrossRef CAS.
- X. F. Huang, M. Salman and Z. Z. Huang, Chem. – Eur. J., 2014, 20, 6618–6621 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Additional optimisation and control reactions, DOE results, experimental procedures and characterisation data. See DOI: 10.1039/d0cc01576h |
| This journal is © The Royal Society of Chemistry 2020 |