
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Cell-penetrating peptides containing 2,5-diketopiperazine (DKP) scaffolds as shuttles for anti-cancer drugs: conformational studies and biological activity†
Lucia
Feni‡
a,
Linda
Jütten‡
 b,
Sara
Parente
c,
Umberto
Piarulli
*c,
Ines
Neundorf
*a and
Dolores
Diaz
*b
b,
Sara
Parente
c,
Umberto
Piarulli
*c,
Ines
Neundorf
*a and
Dolores
Diaz
*b
aUniversity of Cologne, Department of Chemistry, Biochemistry, Zülpicher Str. 47a, D-50674 Cologne, Germany. E-mail: ines.neundorf@uni-koeln.de
bUniversity of Cologne, Department of Chemistry, Organic Chemistry, Greinstraße 4, D-50939, Cologne, Germany. E-mail: mdiazher@uni-koeln.de
cDipartimento di Scienza e Alta Tecnologia, Università degli Studi dell’Insubria, Via Valleggio 11, 22100, Como, Italy. E-mail: Umberto.Piarulli@uninsubria.it
First published on 16th April 2020
Abstract
A series of linear and cyclic peptidomimetics composed of a cell-penetrating peptide and a non-natural, bifunctional 2,5-diketopiperazine scaffold is reported. Conformational studies revealed well-defined helical structures in micellar medium for linear structures, while cyclic peptidomimetics were more flexible. Biological investigations showed higher membrane-activity of cyclic derivatives allowing their use as shuttles for anti-cancer drugs.
The potential use of peptides as therapeutics has gained significant attention in the last decades, and research about the critical factors that favor the biological activity of naturally occurring peptides has been crucial to ease the design of new synthetic analogues.1–5 Fast biological degradation is one of the shortcomings of peptides, and a number of strategies to decrease proteolysis has been proposed being cyclization a very effective one.6 Naturally occurring cyclic peptides have been actively investigated as potential sources of new drugs and antibiotics.7–14 They are particularly resistant to proteolytic degradation when compared with linear peptide chains, and are, therefore, reliable templates for the design and biological modulation of new peptide therapeutics, including peptide carriers, such as cell-penetrating peptides (CPPs).
CPPs are usually short peptides with a net positive charge at physiological pH, which are able to cross cell membranes and thereby transporting cargos inside cells.5b,15 Cyclization of CPPs has been demonstrated to be an effective strategy for enhancing their metabolic stability, cellular uptake rates and promoting endosomal escape and cytoplasmic distribution.16–19 The positively charged side chains are often forced into maximally distant positions stimulating the interaction with negatively charged constituents of the outer plasma membrane in a controlled manner.20 Recently, we presented triazole-bridged cyclic variants of the CPP sC18*21 having superior biological activities compared to their linear counterparts.22
Now, we have synthesized a novel series of linear and cyclic sC18* peptide derivatives including different diastereomers of a bifunctional 2,5-diketopiperazine (DKP) within the sequence (Table 1). The present study aims to expand our knowledge about the impact of non-natural scaffolds on conformational preferences of sC18* to make it more suited for cellular translocation.
| DKP scaffold | Peptide | Sequence |
|---|---|---|
| a Dau: daunorubicin attached to Lys4via aminooxyacetic acid (Aoa). | ||
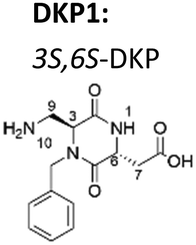
|
1 linear DKP1-sC18* | DKP1-G1LRKRLRKFRNK12 |
| 2 linear DKP3-sC18* | DKP3-GLRKRLRKFRNK | |
| 3 cyclic [DKP1-sC18*] | c(DKP1-GLRKRLRKFRNK) | |
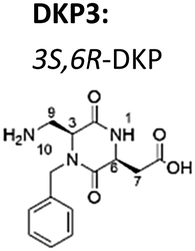
|
4 cyclic [DKP3-sC18*] | c(DKP3-GLRKRLRKFRNK) |
| 5 cyclic[DKP3-sC18*(K4-Aoa-Dau)a] | c(DKP3-GLRK(Dau)RLRKFRNK) | |
| 6 sC18*(K4-Aoa-Dau) | GLRK(Dau)RLRKFRNK | |
The synthesis of the linear peptides 1 and 2 was achieved by manually coupling the DKP scaffolds, synthesized as reported elsewhere,23 to the N-terminus of sC18* while the peptide chain was still immobilized on the solid support. To obtain the cyclic derivatives 3 and 4, cyclization was performed in solution on the crude protected linear peptide after cleavage from the resin (for details see the ESI†). A high dilution was essential to promote the intramolecular reaction, avoiding the formation of dimers. In both cases, however, cyclization remained the major yield-limiting step.
To get detailed information about the crucial structural differences between linear and cyclic DKP-peptidomimetics 1–4, we acquired and analyzed CD and NMR spectra of the synthesized peptides in different media.
CD spectra showed that peptides 1 and 2 were unstructured in phosphate buffer (PBS), while in the presence of 50% trifluoroethanol (TFE) both derivatives showed a positive band at 192 nm and two negative bands at ca. 207 nm and ca. 221 nm indicating the presence of secondary structure motifs (i.e. helix) (see Fig. S7, ESI†). Regarding DKP-cyclic peptides, the CD spectrum of 3 in PBS exhibited a negative band at ca. 200 nm suggesting predominance of structural disorder that was still present in aqueous TFE mixtures. For peptide 4 two very weak negative bands at ca. 204 nm and 225 nm addressed a certain degree of ordered structure in the PBS sample that was tentatively identified as β-hairpin,24 and which was still present when in TFE environment.
Next, we sought to elucidate the 3D structure of peptides 1–4 in both aqueous and membrane mimetic media (i.e. SDS micellar medium) by NMR spectroscopy.
In agreement with CD spectra analysis, the NMR data from linear peptides 1 and 2 in water fitted with unordered structures suggesting, therefore, that the insertion of the DKP scaffold at the N-terminus of the linear peptide sC18* did not suffice to induce secondary structure. Different from that, the spectra of 1 and 2 in micellar medium confirmed the formation of α-helices containing two turns (L2–R10) (see Fig. S9, ESI†). We reasoned further that the positively charged amino acid residues (i.e. Arg and Lys) within the resulting amphipathic helix might facilitate peptide–micelle interactions via electrostatic forces. Interestingly, DKP scaffolds were not involved in the formation and/or stabilization of the helices.25
Regarding the cyclic derivatives 3 and 4, inspection of the amide-aliphatic regions of the 2D 1H,1H-TOCSY spectra in aqueous medium showed that stereochemistry of the DKP linker influenced the diastereotopicity of the methylene protons of G1 and DKP of the trans-derivative 4, but had not such an effect on the signals of 3 (see Fig. S10, ESI†). For both cyclic peptides, the amide signal of F9 is significantly shifted to the lowest frequency region. The amide signal of G1 of 3, however, is remarkably unshielded, while for peptide 4 three signals are shifted at the highest range (i.e.DKP3, G1 and N11) indicating, therefore, that peptides conformations of 3 and 4 were different and depended on the stereochemistry of the DKP scaffold.
In the 2D NOE spectra of peptides 3 and 4 cross-peaks Hα(K12)-H10(DKP) and H7(DKP)-Hα(G1) confirmed covalent connectivity between K12-DKP-G1 proving that the cycle was formed. We also confirmed the stereochemistry of the DKP scaffolds by identification of unambiguous intra-residual NOE cross-peaks for both cis- and trans-DKP derivatives (i.e. H7–H9 for DKP1 and H6–H9 for DKP3). A small number of inter-residual NOEs was collected at r.t. and, consequently, the final ensemble of calculated structures did not converge in a single conformation reflecting, therefore, a rapid equilibrium between several conformational families in solution (see Fig. S11, ESI†).
As the biological testing gave reasons to explore the latter hypothesis (vide infra), we performed variable temperature NMR experiments (see Tables S1–S4, ESI†) to explore whether the most stable conformation would be favored and detectable at low temperature. At 283 K inter-residual amide–amide NOE cross-peaks characteristic of ordered structures were detected supporting the notion that for the cyclic peptides 3 and 4 several conformations co-exist in solution in rapid exchange including, possibly, also a proportion of random coil structures. Interestingly, the number and identity of the amide–amide NOE contacts for cis- and trans cyclic DKP-derivatives were not coincident suggesting that different conformational preferences were also influenced by the stereochemistry of the DKP scaffold. The data fitted with ordered segments within the peptide structure and, indeed, fragments of 3 (i.e. K8F9R10 and DKP1-G1L2) and 4 (i.e. R10N11K12-DKP3 and L2R3) seemed to adopt ordered backbone conformations (see Fig. S12, ESI†).
We performed a molecular modelling study to identify peptides conformers that would fit with the experimental data of 3 and 4 (ESI†). Thus, we first generated random conformations of the peptides and used them as starting geometries for the unrestrained mixed-mode Metropolis Monte Carlo/Stochastic dynamics (MC/SD) simulations with an OPLS_2005 force field. In Fig. 1 the pattern of hydrogen-binding probability deduced from all calculated structures of 4 is shown. All calculated structures exhibited a minimal β-turn motif between the four adjacent amino acids F9–K12 (i.e. (C2O)K12-(NH)F9 and (C2O)F9-(NH)K12). The DKP3 scaffold itself also participated in the formation of hydrogen-bonds so that a turn-like structure is further promoted (i.e. (C2O)DKP3-(NH)R7). For the G1–R5 moiety the probabilities for the formation of inter-residual hydrogen bonds varied between clusters supporting again the assumption that multiple structural families were present in solution because of the flexibility associated to this fragment (vide supra). For the purpose of comparison, a representative NMR derived structure of 4 and a closer view of the fragment K8–K12 are also shown. Identical calculations were carried out with peptide 3 and we concluded that 3 is relatively more unstructured than 4 (see Fig. S13, ESI†).
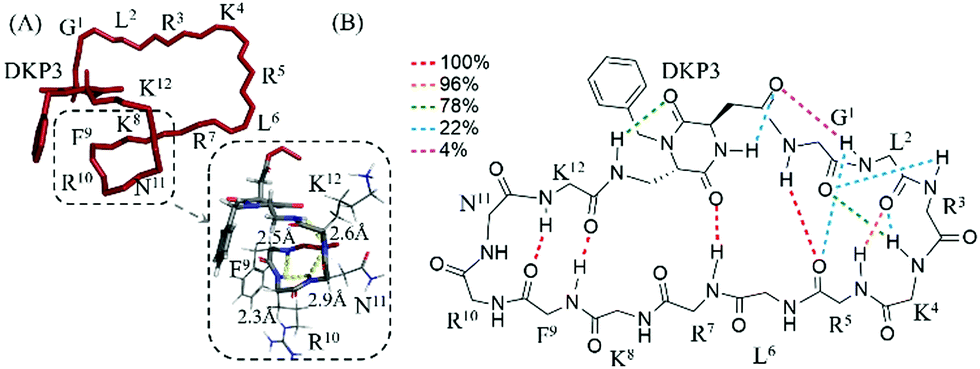 | ||
| Fig. 1 (A) Representative NMR derived structure of 4 in water at 283 K. (B) Intramolecular hydrogen-bonded pattern proposed for the calculated structures of 4. The probability of occurrence of the hydrogen bonds (dotted lines) is color-coded as indicated in the legend (e.g. in 100% of total calculated structures hydrogen bonding between (C2O)K12-(NH)F9 and (C2O)F9-(NH)K12 was observed). | ||
Lastly, we studied the interaction of 3 and 4 with anionic SDS micelles. The signals in the 1D 1H NMR spectra in micellar medium were noticeably broader supporting a strong peptide–micelle interaction and suggesting that peptides might be located not only on the external layer but also embedded into the micelle.
The diffusion coefficient values of 3 and 4 in aqueous medium and in the presence of SDS micelles were compared and the differences correlated with an effective interaction between both cyclic peptides and SDS micelles (see Fig. S14, ESI†).
The assignment of the spectra was hindered by poor signal resolution and severe signal overlapping and we only succeeded with the assignment of 3 in the presence of SDS micelles, being the assignment of 4 unfeasible. The 2D NOESY spectra of 3 acquired at 298 K contained a number of amide–amide cross peaks indicating that the equilibrium between multi-conformers in micellar medium favored specific conformations that are, apparently, less flexible to better interact with the micellar layer (Fig. 2). The arrangement of positively charged side chains (i.e. Arg and Lys) suggested that region G1–R7 may be involved in the interaction with the anionic micelles.
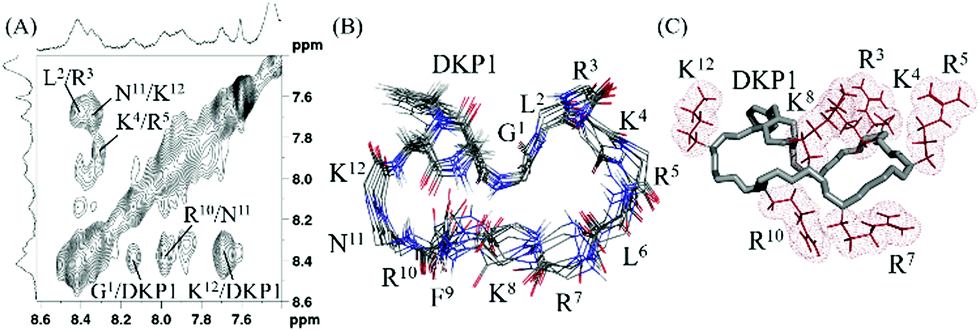 | ||
Fig. 2 (A) Amide–amide region of the 2D 1H,1H-NOESY spectra of 3 in presence of SDS micelles (50 mM phosphate buffer, pH 6.08, water![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :D2O 9:1, 283 K, mixing time 200 ms, 600 MHz). (B) Backbone superimposition of the ensemble of 10 final energy-minimized structures derived from experimental data. (C) Charged side chains (i.e. Arg and Lys) are positioned mainly in one side of the peptide to possibly facilitate interaction with SDS anionic micelles. :D2O 9:1, 283 K, mixing time 200 ms, 600 MHz). (B) Backbone superimposition of the ensemble of 10 final energy-minimized structures derived from experimental data. (C) Charged side chains (i.e. Arg and Lys) are positioned mainly in one side of the peptide to possibly facilitate interaction with SDS anionic micelles. | ||
To summarize, the experimental data reported thus far suggested that the three-dimensional structure of both linear and cyclic DKP-peptidomimetics depended on the properties of the surrounding medium. Accordingly, while linear derivatives formed α-helices in the presence of SDS micelles, the cyclic derivatives exhibited a tendency to form β-turns. Moreover, our studies allow the conclusion that cyclic peptides arrange their positively charged side chains in the direction of the external layer of the micelles which facilitates their interaction with lipids of the cellular membrane. Therefore, in the second part of this work, we were eager to learn more about the biological performance of this novel DKP-peptide series.
We first evaluated the cytotoxicity of compounds 1–4 on HeLa cells (see Fig. S15A, ESI†). After 72 h, the cellular toxicity was in the same range for both linear and cyclic derivatives at low concentrations, but at the highest concentration cyclic peptides were significantly more toxic than their linear counterparts. This observation could be consequence of a more efficient interaction of cyclic peptides with cellular membranes, what would be in perfect agreement with the NMR data, where a more efficient interaction between cyclic DKP-peptides and SDS micelles was underpinned. In addition, also DKP stereochemistry may be relevant, since the difference in toxicity between 3 and 4 (at 100 μM) was significant. However, we concluded that all compounds could be safely used for further experiments at lower concentrations, where cells were almost not harmed. Following, we studied peptide 4 in comparison to its linear version 2 in more detail, since our data reported thus far led us to hypothesize that 4 might be more membrane-active compared to 3.
Since cyclization is known to limit proteolytic degradation, we analyzed the stability of 2 and 4 in the presence of trypsin and cell culture supernatant. Notably, stability of both peptides after treatment with trypsin was very low and no parent peptide was visible after 5 minutes (see Fig. S16A, ESI†). The intrinsic flexibility of both linear 2 and cyclic 4 may support their interaction with the proteolytic enzyme. Remarkably, however, we observed that longer fragments originating from 4 were still detectable after 30 minutes. (i.e. F9R10N11K12-DKP3-G1L2R3K4), while the dominant peak from 2 after 15 minutes. was considerably shorter (i.e.DKP3-G1L2R3).
The same experiments using cell culture supernatant demonstrated that degradation of peptides 2 and 4 was slower, but comparable fragments to the previous experiment were formed. From this we suggested different trypsin interacting moieties for linear and cyclic derivatives, and possibly higher toxicity of cyclic peptides owing to the remaining activity of their lengthier fragments (see Fig. S16B, ESI†).
Finally, we probed if the peptides were able to support the intracellular uptake of a cytostatic drug (e.g. daunorubicin) by non-covalent interactions between cargo and peptide. We chose daunorubicin, because, together with its hydroxy-derivative doxorubicin, it is an often-used drug for such proof-of-principle studies.26 Cellular uptake of daunorubicin in HeLa cells was clearly preferred and increased by up to 25% in the presence of peptides 2 and 4, possibly because the interaction of the peptides with cell membranes facilitated internalization of the drug (see Fig. S15B, ESI†). Cell viability studies using HeLa cells did not show, however, any increase of toxic activity for the mixture of both agents when compared with the drug alone (see Fig. S15C, ESI†).
To see if we could enhance the biological activity, we decided to covalently couple the drug to the side chain of K4 of cyclic compound 4 resulting in compound 5 (Table 1 and ESI†). For reasons of comparison, we also synthesized the linear peptide 6 (i.e. sC18*–Dau). In both cases Dau was coupled via an aminooxyacetic (Aoa) acid linker to the lysine side chain of the peptides. After 30 minutes of incubation in HeLa cells, both peptide-conjugates, 5 and 6, were distributed throughout the cytosol as well as the nuclei (Fig. 3A). For derivative 5 the uptake seemed to be more efficient, and this was quantitatively confirmed by flow cytometry (Fig. 3B). Of more interest was that cell viability studies with compounds 5 and 6 demonstrated higher efficiency of conjugate 5 compared to the linear one, with a more than twofold increased IC50 value (Fig. 3C). The reason for this might be the already discussed more intensive membrane interaction of 4.
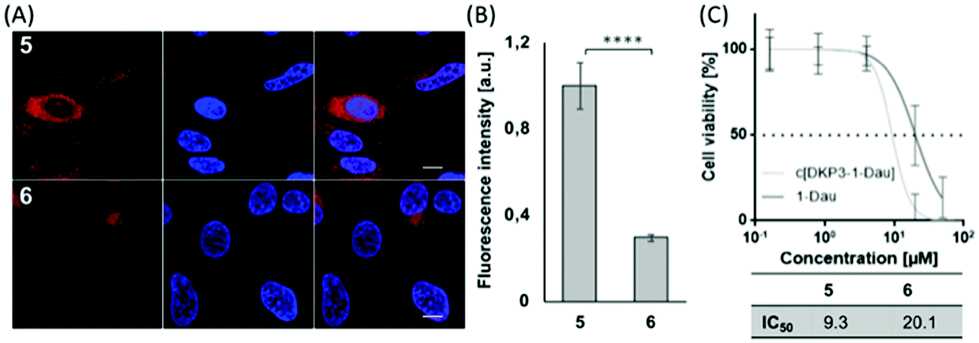 | ||
| Fig. 3 (A) Cellular uptake of 5 and 6 (5 μM) in HeLa cells measured by CLSM and (B) by FACS (10 μM). (C) Cell viability assay of compounds 5 and 6. Cells were treated as described in Fig. S15A (ESI†). ***p ≤ 0.001. | ||
To conclude, we have presented a novel group of linear and cyclic cell-penetrating peptides containing bifunctional diketopiperazine scaffolds (DKP1 and DKP3) with a single difference regarding stereochemistry of one of the stereocenters of the scaffold. Conformational studies of the synthesized peptides were carried out by a combination of CD and NMR spectroscopy and are supported by molecular modelling. We have demonstrated that the synthetic DKP–sC18* peptidomimetics showed a clear conformational switch when in contact with membrane mimetic agents. Moreover, cyclic variants were characterized by arranging their positively charged residues to the hydrophobic phase in a more rigid conformation presumably favoring membrane interaction. This conclusion matched very well to our results from the cell viability studies, where both linear and cyclic derivatives exhibited comparable cytotoxic profiles at relatively low concentrations, but where a noticeable detriment for the cyclic peptides at high concentrations was detected. This might be taken as an indication of a divergent mode of interaction with the cellular membranes and a better stability upon treatment with proteases, what was also measured for the cyclic version 4. Furthermore, cyclic derivative 4 was proven as more efficient drug transporter compared to recently reported linear version sC18*, which indicated that covalent coupling might be a preferred strategy. Taken together, these results suggest that we have generated new drug delivery systems with high penetration ability, greater stability to protease degradation and ability to transport a cargo into the cell. In the future, we will study in more detail the cellular uptake of those cyclic peptides and if they can be used for anti-tumoral cargo delivery in vivo.
The European Commission (MSCA-ITN MAGICBULLET 642004) supported this work. We acknowledge the CECAD Imaging Facility of University of Cologne for assistance with the generation of microscopy data. D. D. is grateful to the University of Cologne for supporting the JobSharing project.
The NMR equipment used for this work (600 MHz) was funded by a ‘HBFG’ grant (HBFG 111/602-1) of the Federal State of North Rhine-Westphalia and the Deutsche Forschungsgemeinschaft (DFG).
Conflicts of interest
There are no conflicts to declare.Notes and references
- J. L. Lau and M. K. Dunn, Bioorg. Med. Chem., 2018, 26, 2700 CrossRef CAS PubMed.
- V. Vergote, C. Burvenich, C. Van de Wiele and B. De Spiegeleer, J. Pept. Sci., 2009, 15(11), 697–710 CrossRef CAS PubMed.
- P. Wójcik and L. Berlicki, Bioorg. Med. Chem. Lett., 2016, 26, 707–713 CrossRef PubMed.
- A. A. Bahar and D. Ren, Pharmaceuticals, 2013, 6(12), 1543–1575 CrossRef PubMed.
- (a) K. Hoffmann, N. Milech, S. M. Juraja, P. T. Cunningham, S. R. Stone, R. W. Francis, M. Anastasas, C. M. Hall, T. Heinrich, H. M. Bogdawa, S. Winslow, M. N. Scobie, R. E. Dewhurst, L. Florez, F. Ong, M. Kerfoot, D. Champain, A. M. Adams, S. Fletcher, H. M. Viola, L. C. Hool, T. Connor, B. A. C. Longville, Y. F. Tan, K. Kroeger, V. Morath, G. A. Weiss, A. Skerra, R. M. Hopkins and P. M. Watt, Sci. Rep., 2018, 8(1), 12538 CrossRef PubMed; (b) D. Kalafatovic and E. Giralt, Molecules, 2017, 22(11), 1929 CrossRef PubMed.
- S. Sachdeva, Int. J. Pept. Res. Ther., 2017, 23, 49–60 CrossRef CAS.
- G. F. Gause, Lancet, 1946, 2(6411), 46 CrossRef CAS.
- S. H. Joo, Biomol. Ther., 2012, 20(1), 19–26 CrossRef CAS PubMed.
- (a) G. G. Newton and E. P. Abraham, Biochem. J., 1953, 53(4), 597–604 CrossRef CAS PubMed; (b) W. A. Toscano, Jr. and D. R. Storm, Pharmacol. Ther., 1982, 16(2), 199–210 CrossRef.
- T. Velkov, K. D. Roberts, R. L. Nation, P. E. Thompson and J. Li, Future Microbiol., 2013, 8(6), 711–724 CrossRef CAS PubMed.
- A. Laupacis, P. A. Keown, R. A. Ulan, N. McKenzie and C. R. Stiller, Can. Med. Assoc. J., 1982, 126(9), 1041–1046 CAS.
- G. Moldenhauer, A. V. Salnikov, S. Lüttgau, I. Herr, J. Anderl and H. Faulstich, J. Natl. Cancer Inst., 2012, 104(8), 622–634 CrossRef CAS PubMed.
- M. A. Abdalla and L. J. McGaw, Molecules, 2018, 23(8), 2080 CrossRef PubMed.
- H. Hashizume, R. Sawa, K. Yamashita, Y. Nishimura and M. Igarashi, J. Antibiot., 2017, 70(5), 699–704 CrossRef CAS PubMed.
- K. Kardani, A. Milani, S. H. Shabani and A. Bolhassani, Expert Opin. Drug Delivery, 2019, 16(11), 1227–1258 CrossRef CAS PubMed.
- Z. Qian, T. Liu, Y. Y. Liu, R. Briesewitz, A. M. Barrios, S. M. Jhiang and D. Pei, ACS Chem. Biol., 2013, 8(2), 423–431 CrossRef CAS PubMed.
- L. Cascales, S. T. Henriques, M. C. Kerr, Y. H. Huang, M. J. Sweet, N. L. Daly and D. J. Craik, J. Biol. Chem., 2011, 286(42), 36932–36943 CrossRef CAS PubMed.
- D. Mandal, A. Nasrolahi Shirazi and K. Parang, Angew. Chem., Int. Ed., 2011, 50(41), 9633–9637 CrossRef CAS PubMed.
- H. Traboulsi, H. Larkin, M. A. Bonin, L. Volkov, C. L. Lavoie and É. Marsault, Bioconjugate Chem., 2015, 26(3), 405–411 CrossRef CAS PubMed.
- G. Lättig-Tunnemann, M. Prinz, D. Hoffmann, J. Behlke, C. Palm-Apergi, I. Morano, H. D. Herce and M. C. Cardoso, Nat. Commun., 2011, 2, 453 CrossRef PubMed.
- M. Horn, F. Reichart, S. Natividad-Tietz, D. Díaz and I. Neundorf, Chem. Commun., 2016, 52(11), 2261–2264 RSC.
- (a) M. Horn and I. Neundorf, Sci. Rep., 2018, 8(1), 16279–16290 CrossRef PubMed; (b) A. Gronewold and I. Neundorf, Beilstein J. Org. Chem., 2018, 14, 1378–1388 CrossRef CAS PubMed.
- M. Marchini, M. Mingozzi, R. Colombo, I. Guzzetti, L. Belvisi, F. Vasile, D. Potenza, U. Piarulli, D. Arosio and C. Gennari, Chem. – Eur J., 2012, 18(20), 6195–6207 CrossRef CAS PubMed.
- A. C. Gibbs, L. H. Kondejewski, W. Gronwald, A. M. Nip, R. S. Hodges, B. D. Sykes and D. S. Wishart, Nat. Struct. Mol. Biol., 1998, 5, 284–288 CrossRef CAS PubMed.
- L. Vahdati, J. Kaffy, D. Brinet, G. Bernadat, I. Correia, S. Panzeri, R. Fanelli, O. Lequin, M. Taverna, S. Ongeri and U. Piarulli, Eur. J. Org. Chem., 2017, 2971–2980 CrossRef CAS.
- (a) L. Feni, S. Parente, C. Robert, S. Gazzola, D. Arosio, U. Piarulli and I. Neundorf, Bioconjugate Chem., 2019, 30(7), 2011–2022 CrossRef CAS PubMed; (b) A. Accardo, S. Mannucci, E. Nicolato, F. Vurro, C. Diaferia, P. Bontempi, P. Marzola and G. Morelli, Drug Delivery Transl. Res., 2019, 9(1), 215–226 CrossRef CAS PubMed; (c) A. A. P. Tripodi, I. Ranőelović, B. Biri-Kovács, B. Szeder, G. Mező and J. Tóvári, Pathol. Oncol. Res., 2019 DOI:10.1007/s12253-019-00773-3.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0cc01490g |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2020 |