
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceC–F activation reactions at germylium ions: dehydrofluorination of fluoralkanes†
Maria
Talavera
 ,
Gisa
Meißner
,
Simon G.
Rachor
and
Thomas
Braun
*
,
Gisa
Meißner
,
Simon G.
Rachor
and
Thomas
Braun
*
Department of Chemistry, Humboldt-Universität zu Berlin, Brook-Taylor-Str. 2, 12489 Berlin, Germany. E-mail: thomas.braun@cms.hu-berlin.de
First published on 20th March 2020
Abstract
Reactions of the trityl cations with germanes afford the germylium ions [R3Ge][B(C6F5)4] (1a: R = Et, 1b: R = Ph, 1c: R = nBu). These compounds react with germane or fluorogermane to give polynuclear species, which are sources of the mononuclear ions, The latter convert with phosphines to yield the [R3Ge-PR3]+ (4a: R = Et, 4b: R = Ph) cations. Catalytic dehydrofluorination reactions were observed for the C–F bond activation of fluoroalkanes when using germanes as hydrogen source.
Fluorinated compounds play an important role in everyday life in terms of agrochemicals, pharmaceuticals, liquid crystals and cooling agents.1 Catalytic C–F activation reactions to access fluorinated building blocks open up opportunities in synthetic chemistry, but can often be considered as a major challenge,2 which is frequently overcome by the formation of strong H–F, B–F, Al–F, Si–F or Ge–F bonds.2,3 In this regard, strong Lewis-acidic main-group compounds such as silylium ions4 were used to induce C–F activation reactions resulting often in hydrodefluorinations in the presence of hydrogen sources.
During the last years, germylium ions4g,5 have been obtained via different synthetic approaches. They often bear bulky substituents and are stabilized by weakly coordinating anions. One of the most used methodologies consists of a hydride transfer reaction at germanes to a trityl cation. Thus, in 2013 Müller et al. described the syntheses of sterically bulky germylium ions such as [Mes3Ge]+, where the latter is formed from Mes2MeGeH by a rearrangement reaction.4g One year later, they synthesized a hydrogen bridged naphtyldigermylium ion capable of performing catalytic hydrodefluorination reactions at C(sp3)–F bonds using Et3SiH as hydrogen source with comparable turnover numbers than those found for similar silylium ions as catalyst.5i Catalytic hydrodefluorination reactions have also been recently published by Weinert and co-workers using in situ formed [Ph3Ge][B(C6F5)4] ions in neat substrates.6 However, literature known trialkyl germylium ions are limited to [R3Ge]+ (R = Me, Et) stabilized by carborane anions and no studies on bond activation reactions are known.5d,g In general, investigations on the reactivity of germylium ions towards organic compounds are very scarce5i,l,n,o,6 and so far, stoichiometric C–F bond activation reactions have not been studied.
Herein, we describe the generation and identification of the germylium ions [R3Ge]+ with [B(C6F5)4]− as counteranion (1a: R = Et, 1b: R = Ph, 1c: R = nBu) and their role in the C–F bond activation reactions of fluorinated alkanes. The reactions led not to hydrodefluorination reactions, but instead to unprecedented dehydrofluorination reactions at molecular main group compounds.
Treatment of [Ph3C][B(C6F5)4] with one equivalent of triethyl-, triphenyl- or tributylgermane gave [R3Ge][B(C6F5)4] (1a: R = Et, 1b: R = Ph, 1c: R = nBu) as well as Ph3CH (Scheme 1). NMR spectroscopic data confirmed the consumption of the corresponding germane due to the abstraction of a hydride by the trityl cation. In the case of the cation in 1a, the quartet for the CH2 moiety appears Δδ = 0.5 ppm shifted towards higher field with respect to the previously synthesized [Et3Ge][CHB11H5Br6] germylium ion.5g LIFDI-TOF mass spectra show the isotopic pattern and molar masses of [R3Ge]+ (see ESI†).
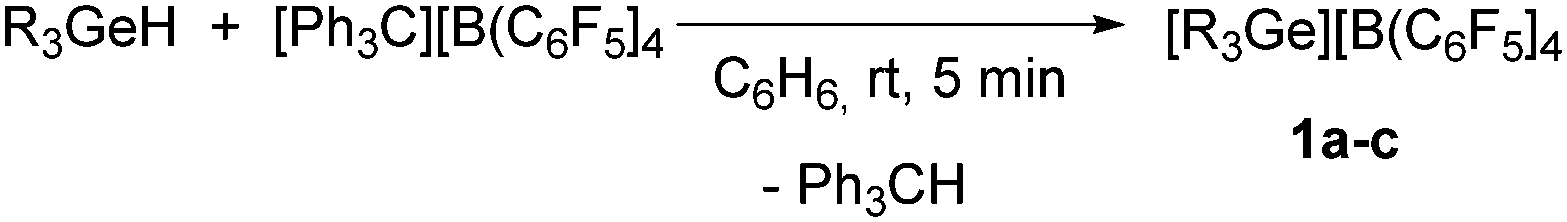 | ||
| Scheme 1 Formation of the germylium ions 1 (1a: R = Et, 1b: R = Ph, 1c: R = nBu). | ||
As it has been previously reported by Reed et al. that with carboranes as counteranions, digermylium ions might be generated in the presence of an excess of germane.5g On treatment of [R3Ge][B(C6F5)4] (1a–c) with one equivalent of germane the formation of polynuclear species such as the digermylium ions [R3Ge–H–GeR3][B(C6F5)4] (2a–c) can be assumed (Scheme 2). Broad signals at δ = 2.08 ppm (for R = Et), δ = 5.40 ppm (for R = Ph) and δ = 1.97 ppm (for R = nBu) in the 1H NMR spectra can be assigned to the proton bridging two germylium centres. In every case, the resonance appears shielded with respect to R3GeH (Δδ = 1.88, 0.55 and 2 ppm for R = Et, R = Ph and R = nBu, respectively), which is in accordance with data for the previously described naphtyldibutyldigermylium cation.5i However, a DOSY NMR experiment of a product mixture of compound 1a and Et3GeH indicated that more than one species are present. Note that after adding more Et3GeH to the mixture of 1a and Et3GeH a shift of the 1H NMR signal to lower field as well as the formation of GeEt4 was observed, which suggest the occurrence of rearrangement reactions.4g
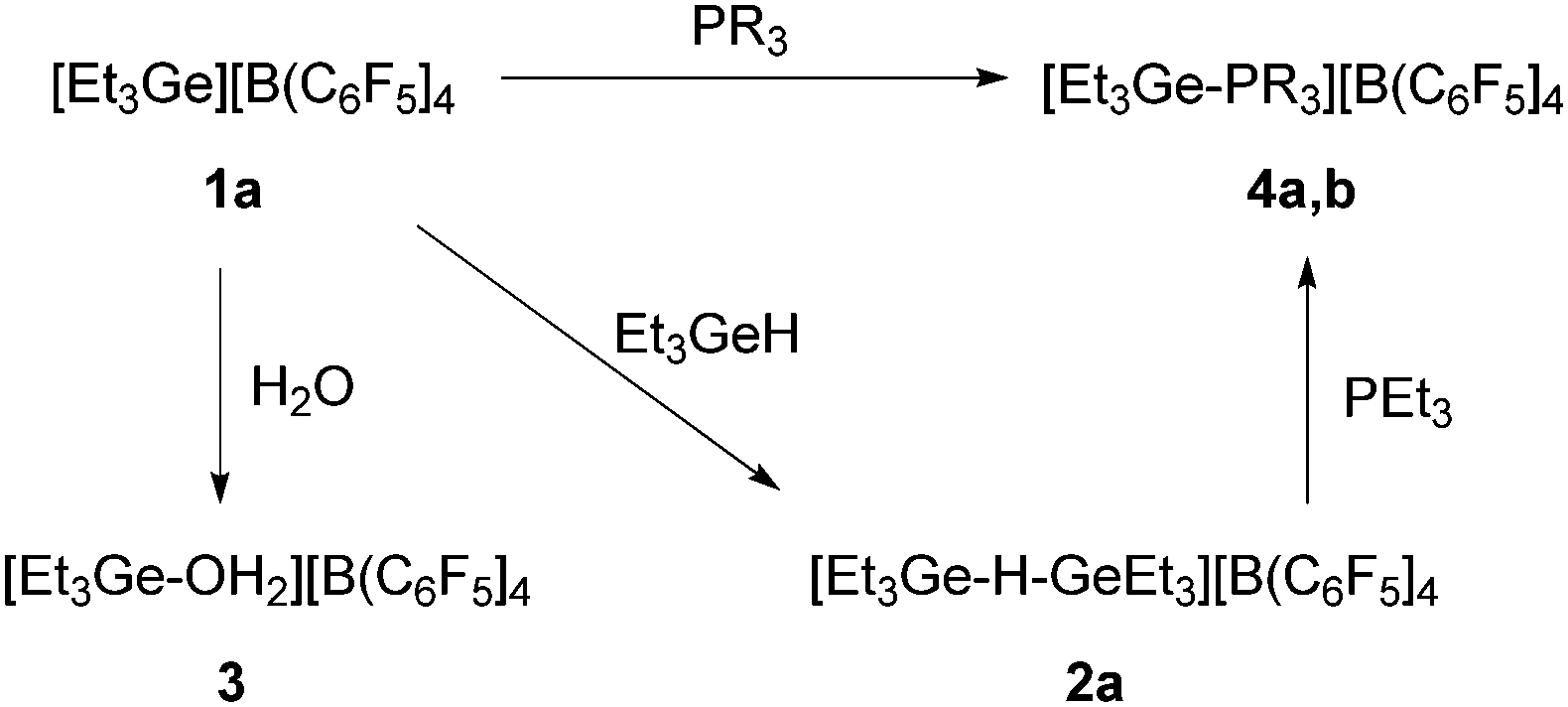 | ||
| Scheme 2 Reactivity of 1a. All reactions were performed in ortho-dichlorobenzene-d4 at room temperature for 5 min (4a: R = Et, 4b: R = Ph). | ||
The germylium ions in 1 are highly water-sensitive. Thus, 1a reacts with water to yield [Et3Ge–OH2][B(C6F5)4] (3) (Scheme 2). The 1H NMR spectrum of 3 reveals a very broad signal at 5.1 ppm due to the protons at the germanium bound water. The molecular structure of compound 3 was determined by single-crystal X-ray diffraction analysis (Fig. 1). Compound 3 crystallizes in a distorted tetrahedral structure. The sum of the C–Ge–C angles is 347.26°, which is consistent with literature-known structures for germanols or cations exhibiting hydroxo-bridged germanium centres.5b,7 The Ge–O bond length in 3 of 1.923(2) Å is slightly longer than the distance in the hydroxo digermylium ion [(Me3Ge)2OH]+ (1.897(4) and 1.903(4) Å)5b and around 0.15 Å longer than the Ge–O bond in Ph3GeOH7 or (Ph3Ge)2O.8 Similar differences have been found for silicon analogues.9 Additionally, the asymmetric unit shows a water molecule which binds via a hydrogen bond to the coordinated water molecule with a O–O separation of 2.492 Å.
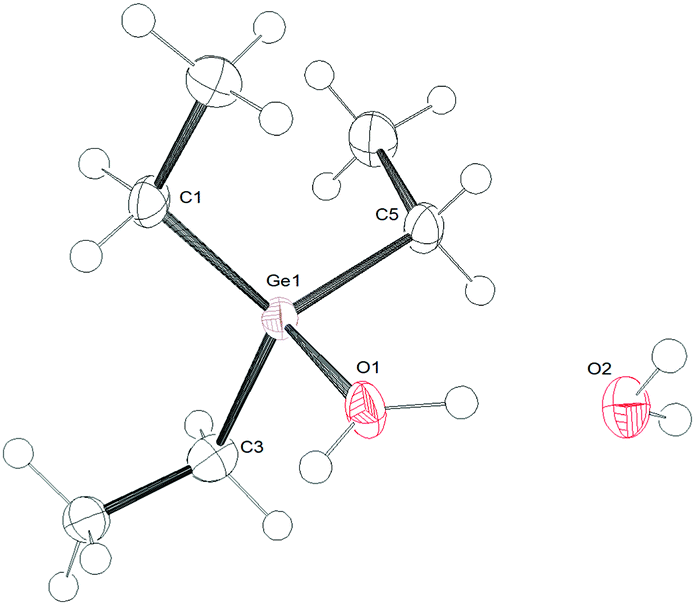 | ||
| Fig. 1 ORTEP representation of the cation in 3·H2O; ellipsoids are drawn at a 50% probability level. Selected bonds lengths (Å) and angles (°): Ge1–O1 1.9247(14); Ge1–C1 1.932(2); Ge1–C3 1.9324(19); Ge1–C5 1.932(2); C3–Ge1–C5 116.42(9); C1–Ge1–C5 113.97(9); C1–Ge1–C3 116.89(9); C3–Ge1–O1 103.43(8); C5–Ge1–O1 101.47(8); C1–Ge1–O1 101.20(8). | ||
In order to get more insight on the structure and reactivity of 1a, it was also reacted with PEt3 or PPh3 in deuterated ortho-dichlorobenzene. After 5 minutes, signals at 1.9 ppm and 2.3 ppm for [Et3Ge–PR3][B(C6F5)4] (R = Et (4a), Ph (4b)) in the 31P{1H} NMR spectra were observed (Scheme 2). Further support for the presence of these compounds was provided by the LIFDI-TOF spectrum of 4b, which reveals a peak at m/z 423 with the corresponding isotopic pattern for the cation (see ESI†).
The mixture of 1a and Et3GeH was also treated with one equivalent of PEt3 (Scheme 2). Again, compound 4a was obtained together with Et3GeH, GeEt4 and Et2GeH2 in an approximate ratio of 8![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2.4:1. This again indicates the occurrence of rearrangement reactions, but also that a mixture of 1 and germanes shows a comparable reactivity to those of 1.
:2.4:1. This again indicates the occurrence of rearrangement reactions, but also that a mixture of 1 and germanes shows a comparable reactivity to those of 1.
Treatment of [R3Ge][B(C6F5)4] (1a: R = Et, 1c: nBu) with fluorocyclohexane in a 1:1 ratio in deuterated 1,2-dichlorobenzene gave the Friedel–Crafts product, 1,2-dichloro-4-cyclohexylbenzene-d3.10 In addition, a broad peak at δ = −200 ppm in the 19F NMR spectrum was observed, which might be assigned to polynuclear species such as [R3Ge–F–GeR3][B(C6F5)4] (5), although a second fluorine containing product was not detected. However, the chemical shift in the 19F NMR spectrum slightly varies over time or after addition of further fluorocyclohexane suggesting the formation of various fluoride-bridge germylium compounds. Additionally, the reaction of a mixture of 1a,c with R3GeH and fluorocyclohexane provided the same result (Scheme 3), giving a signal in the 19F NMR spectrum of the product reaction solution at δ = −196.9 ppm (R = Et) or δ = −201.6 ppm (R = nBu). Note that HD and H2 formation was observed in the 1H NMR spectrum. The presence of fluoride-bridged cations is further supported by a reaction of the compounds 1a,b with R3GeF (R = Et, Ph), which resulted in resonances at δ = −196.9 ppm for R = Et and δ = −170.4 ppm for R = Ph, which appear at lower field than the signals for R3GeF in the 19F NMR spectra (Scheme 3).
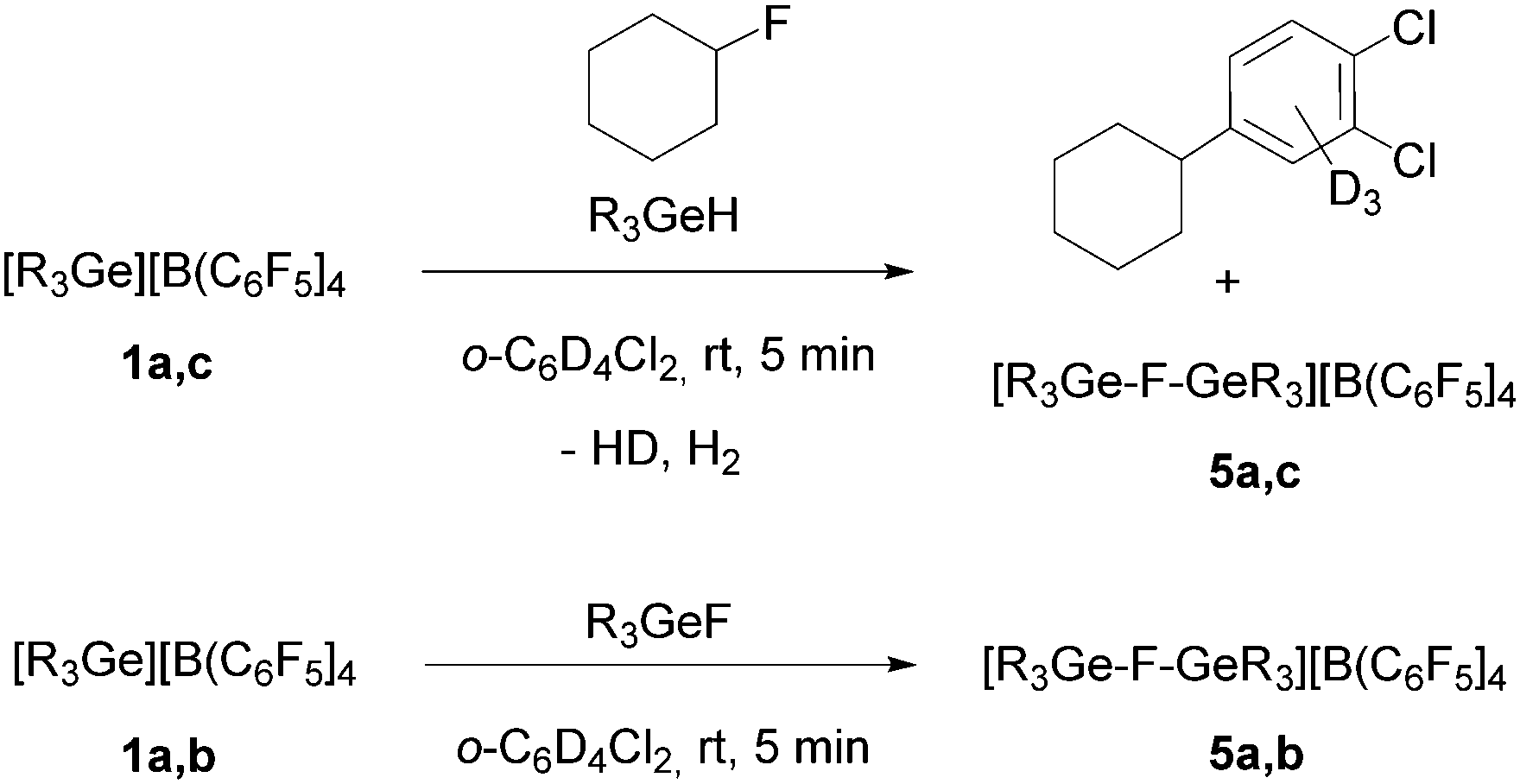 | ||
| Scheme 3 Formation of the germylium ions 5 (5a: R = Et, 5b: R = Ph, 5c: R = nBu). | ||
The in situ generated ions 1 were then used as catalysts (5 mol%) for the C–F bond activation of 1-fluorocyclohexane. In a very different outcome, the presence of two equivalents of Et3GeH or nBu3GeH in deuterated 1,2-dichlorobenzene as solvent promoted the formation of the dehydrofluorinated product cyclohexene (Table 1) together with the corresponding fluorogermane species and dihydrogen. The formation of small amounts of cyclohexane could also be detected. A reaction of Et3GeH or nBu3GeH with fluorocyclohexane in presence of [Ph3C]+ without using a solvent did not lead to any different outcome. The observations are in sharp contrast to the mentioned report on catalytic hydrodefluorination reactions at fluoroalkanes, which occur in the presence of HGePh3 without solvent or by using a naphtyldigermylium ion and silanes as hydrogen source.5i,6 Note that when 1a was used as catalyst and HSiEt3 as the hydrogen source, only hydrodefluorination of fluorocyclohexane is observed, which is consistent with the reactivity pattern of silylium ions.4a,b,e,i
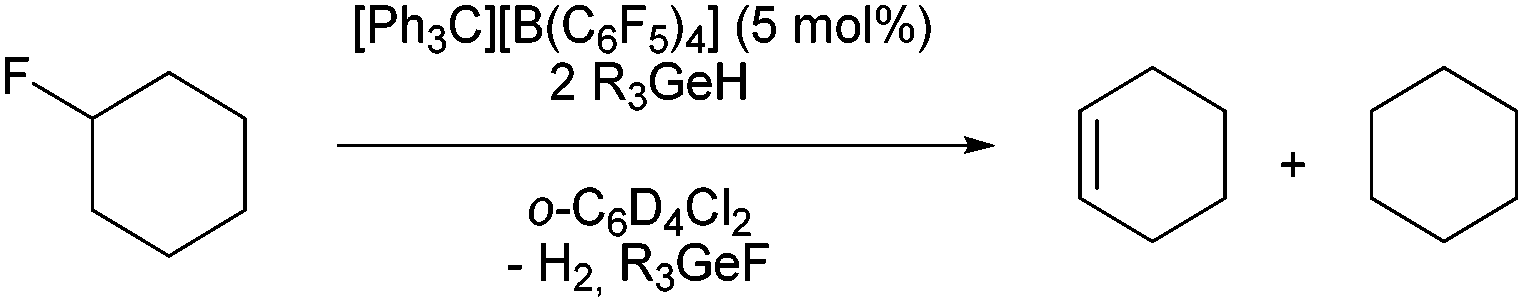
When Et3GeH was used as hydrogen source a TON of 40 could be obtained after five hours. In contrast, nBu3GeH only gave a 23% of conversion at room temperature after four days, however, heating at 65 °C reduced the reaction time to two hours with full conversion of fluorocyclohexane. Note that the triphenylgermylium ions led to reactivity towards the counteranion as well as to the generation of Friedel–Crafts products with the solvent. [Et3Ge-PPh3][B(C6F5)4] (4b) was also tested in the catalytic reaction, which results in a comparable outcome as with [Et3Ge][B(C6F5)4] (1a) when heating at 100 °C. Therefore, triethylgermane was chosen as the best hydrogen source for the following studies, using the trityl cation as pre-catalyst for the in situ formation of the catalyst 1a (Table 2).
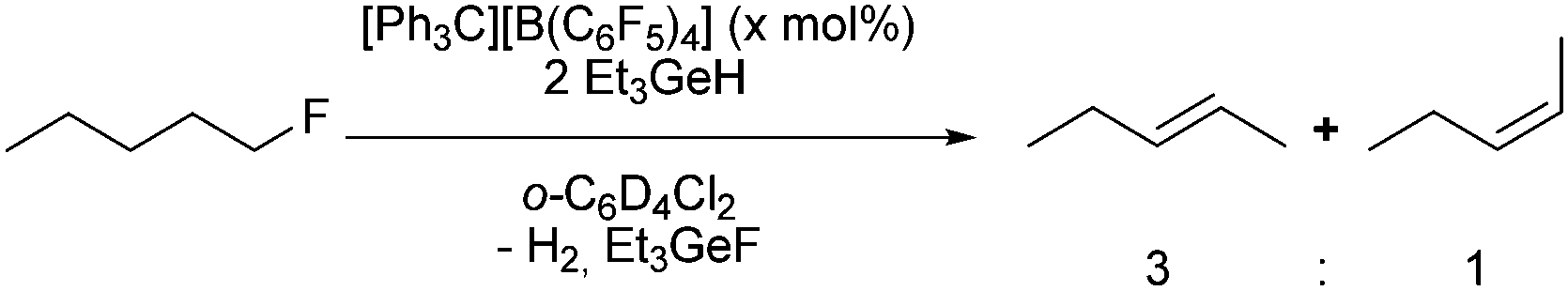
Treatment of a mixture of the trityl cation as catalytic precursor (10 mol%) and triethylgermane as hydrogen source with 1-fluoropentane gave at 65 °C in 3 hours 2-pentene isomers in a ratio of the E- and Z-isomer of 3:1. Other fluorinated compounds such as 1-fluoroheptane and fluoroethane were also tested. Whereas the former gave a mixture of E/Z-2-heptene isomers (3:1 ratio), Et3GeF and dihydrogen, the latter converted into GeEt4, ethane and Et3GeF. Difluoroethane, trifluoroethane and 1,1,1,2,3,3-hexafluoropropane did not react, which suggest that the activation of any CF2 or CF3 moieties is difficult. However, 1,1,1,3,3-pentafluorobutane was converted to 2,4,4,4-tetrafluoro-1-butene in 4.6% after three days at 65 °C.
A tentative mechanistic proposal for the catalytic dehydrofluorination reactions involves the C–F activation of a fluoroalkene by a germylium ion-type species to give fluorogermane and a carbenium-like ion. In the presence of germane, an olefin and dihydrogen are generated in addition to the regeneration of the germylium ion (Scheme 4). The formation of intermediate carbenium-like ions as intermediates is also supported by the formation of 2-pentene isomers instead of 1-pentene when 1-fluoropentane is used as starting material. Note that a mechanism via carbenium-like species based on silylium ions was proposed for hydrodefluorination reactions by Ozerov and Müller,4a,e,i but the presence of germanes in solution obviously favours a dehydrofluorination pathway.
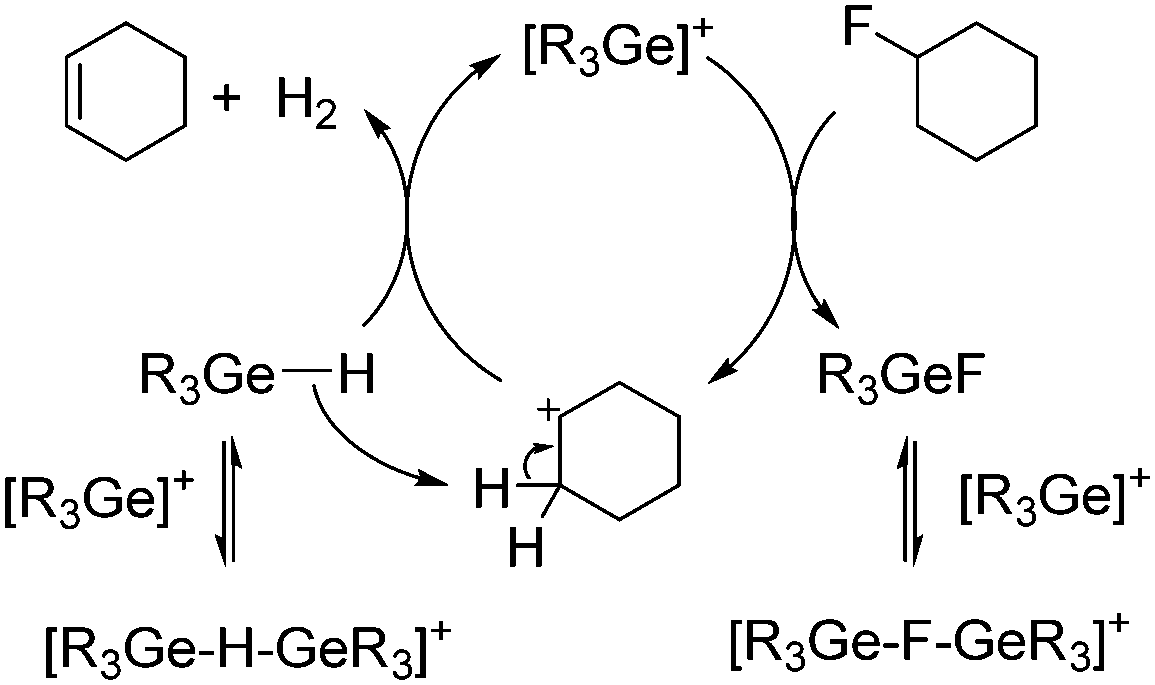 | ||
| Scheme 4 Simplified mechanism of the catalytic dehydrofluorination of fluorocyclohexane by germylium ions (R = Et, nBu). | ||
The germylium ion reactivity resembles the behaviour of germanes in heterogeneous reactions using the Lewis-acid aluminum chlorofluoride ACF (AlClxF3−x, x = 0.05–0.3) as catalyst, for which germylium-type intermediates were proposed, and dehydrofluorination reactions were also found, although 70 °C and 4 d were required.11 Note also that for homogeneous reactions, only very few examples of transition metal complexes of Sc, Ti and Ce have been reported to perform dehydrofluorination reactions via an initial C–H bond activation followed by β-fluoride elimination.12
In conclusion, the synthesis of germylium ions has been achieved and their reactivity towards fluorinated alkanes was studied. Although the structure of the polygermylium ions remains unclear, their reactivity towards phosphines demonstrate their applicability as sources of mononuclear species. The germylium-type ions react with fluoroalkanes to form fluorogermylium ions. When no excess of germane is present, Friedel–Crafts reactivity was observed. However, in the presence of germane the carbenium-like ion reactivity is diminished. Remarkably, catalytic C–F activation at monofluorinated alkanes using Et3GeH or nBu3GeH promote dehydrofluorination reactions instead of the hydrodefluorination products, which occur when silanes are used as hydrogen source. In addition, [Et3Ge]+ or [nBu3Ge]+ favour a reaction pathway towards the dehydrofluorination, although under neat conditions with [Ph3Ge]+ selectivity towards hydrodefluorination was reported.6 Overall, the presented results open opportunities for the development of reactions routes for defluorination of fluorinated alkanes.
G. M. acknowledges the graduate school SALSA (School of Analytical Science Adlershof).
Conflicts of interest
There are no conflicts to declare.Notes and references
- (a) P. Jeschke, ChemBioChem, 2004, 5, 570–589 CrossRef CAS PubMed; (b) P. Kirsch, Modern Fluoroorganic Chemistry: Applications of Organofluorine Compounds, Wiley-VCH, Weinheim, 2nd edn, 2013 CrossRef; (c) K. Müller, C. Faeh and F. Diederich, Science, 2007, 317, 1881–1886 CrossRef PubMed.
- (a) T. G. Richmond, Angew. Chem., Int. Ed., 2000, 39, 3241–3244 ( Angew. Chem. , 2000 , 112 , 3378–3380 ) CrossRef CAS; (b) T. Braun and R. N. Perutz, Chem. Commun., 2002, 2749–2757 RSC; (c) S. A. Macgregor, Chem. Soc. Rev., 2007, 36, 67–76 RSC; (d) H. Amii and K. Uneyama, Chem. Rev., 2009, 109, 2119–2183 CrossRef CAS PubMed; (e) A. D. Sun and J. A. Love, Dalton Trans., 2010, 39, 10362–10374 RSC; (f) J.-F. Paquin, Synlett, 2011, 289–293 CrossRef CAS; (g) M. F. Kuehnel, D. Lentz and T. Braun, Angew. Chem., Int. Ed., 2013, 52, 3328–3348 ( Angew. Chem. , 2013 , 125 , 3412–3433 ) CrossRef PubMed; (h) P. A. Champagne, Y. Benhassine, J. Desroches and J.-F. Paquin, Angew. Chem., Int. Ed., 2014, 53, 13835–13839 ( Angew. Chem. , 2014 , 126 , 14055–14059 ) CrossRef CAS PubMed; (i) T. Ahrens, J. Kohlmann, M. Ahrens and T. Braun, Chem. Rev., 2015, 115, 931–972 CrossRef CAS PubMed; (j) W. Chen, C. Bakewell and M. R. Crimmin, Synthesis, 2017, 810–821 CAS; (k) C. Bakewell, A. J. P. White and M. R. Crimmin, Angew. Chem., Int. Ed., 2018, 57, 6638–6642 ( Angew. Chem. , 2018 , 130 , 6748–6752 ) CrossRef CAS PubMed; (l) Q. Shen, Y.-G. Huang, C. Liu, J.-C. Xiao, Q.-Y. Chen and Y. Guo, J. Fluorine Chem., 2015, 179, 14–22 CrossRef CAS; (m) T. Ahrens, M. Ahrens, T. Braun, B. Braun and R. Herrmann, Dalton Trans., 2016, 45, 4716–4728 RSC.
- Y. R. Luo, Comprehensive Handbook of Chemical Bond Energies, CRC Press, Boca Raton, 2007 Search PubMed.
- (a) R. Panisch, M. Bolte and T. Müller, J. Am. Chem. Soc., 2006, 128, 9676–9682 CrossRef CAS PubMed; (b) C. Douvris and O. V. Ozerov, Science, 2008, 321, 1188–1190 CrossRef CAS PubMed; (c) R. N. Perutz, Science, 2008, 321, 1168–1169 CrossRef CAS PubMed; (d) H. F. T. Klare and M. Oestreich, Dalton Trans., 2010, 39, 9176–9184 RSC; (e) C. Douvris, C. M. Nagaraja, C.-H. Chen, B. M. Foxman and O. V. Ozerov, J. Am. Chem. Soc., 2010, 132, 4946–4953 CrossRef CAS PubMed; (f) O. Allemann, S. Duttwyler, P. Romanato, K. K. Baldridge and J. S. Siegel, Science, 2011, 332, 574–577 CrossRef CAS PubMed; (g) A. Schäfer, M. Reißmann, S. Jung, A. Schäfer, W. Saak, E. Brendler and T. Müller, Organometallics, 2013, 32, 4713–4722 CrossRef; (h) B. Shao, A. L. Bagdasarian, S. Popov and H. M. Nelson, Science, 2017, 355, 1403–1407 CrossRef CAS PubMed; (i) V. J. Scott, R. Çelenligil-Çetin and O. V. Ozerov, J. Am. Chem. Soc., 2005, 127, 2852–2853 CrossRef CAS PubMed; (j) S. Duttwyler, C. Douvris, N. L. P. Fackler, F. S. Tham, C. A. Reed, K. K. Baldridge and J. S. Siegel, Angew. Chem., Int. Ed., 2010, 49, 7519–7522 ( Angew. Chem. , 2010 , 122 , 7681–7684 ) CrossRef CAS PubMed; (k) T. Stahl, H. F. T. Klare and M. Oestreich, J. Am. Chem. Soc., 2013, 135, 1248–1251 CrossRef CAS PubMed; (l) M. Ahrens, G. Scholz, T. Braun and E. Kemnitz, Angew. Chem., Int. Ed., 2013, 125, 5436–5440 ( Angew. Chem., Int. Ed. , 2013 , 52 , 5328–5332 ) CrossRef.
- (a) P. Antoniotti, P. Benzi, F. Grandinetti and P. Volpe, J. Phys. Chem., 1993, 97, 4945–4950 CrossRef CAS; (b) J. B. Lambert, S. M. Ciro and C. L. Stern, J. Organomet. Chem., 1995, 499, 49–55 CrossRef CAS; (c) J. B. Lambert, Y. Zhao, H. Wu, W. C. Tse and B. Kuhlmann, J. Am. Chem. Soc., 1999, 121, 5001–5008 CrossRef CAS; (d) I. Zharov, T.-C. Weng, A. M. Orendt, D. H. Barich, J. Penner-Hahn, D. M. Grant, Z. Havlas and J. Michl, J. Am. Chem. Soc., 2004, 126, 12033–12046 CrossRef CAS PubMed; (e) Y. Yang, R. Panisch, M. Bolte and T. Müller, Organometallics, 2008, 27, 4847–4853 CrossRef CAS; (f) C. Schenk, C. Drost and A. Schnepf, Dalton Trans., 2009, 773–776 RSC; (g) J. H. Wright, G. W. Mueck, F. S. Tham and C. A. Reed, Organometallics, 2010, 29, 4066–4070 CrossRef CAS; (h) D. L. Myalochkin, T. A. Kochina, D. V. Vrazhnov, V. V. Avrorin and E. N. Sinotova, Radiochemistry, 2010, 52, 99–102 CrossRef CAS; (i) N. Kordts, C. Borner, R. Panisch, W. Saak and T. Müller, Organometallics, 2014, 33, 1492–1498 CrossRef CAS; (j) T. A. Kochina, D. L. Myalochkin, V. V. Avrorin and E. N. Sinotova, Russ. Chem. Bull., 2016, 65, 597–620 CrossRef CAS; (k) A. A. Korlyukov, E. A. Komissarov, E. P. Kramarova, A. G. Shipov, V. V. Negrebetsky, S. Y. Bylikin and Y. I. Baukov, Russ. Chem. Bull., 2016, 65, 2583–2593 CrossRef CAS; (l) H. Fang, H. Jing, A. Zhang, H. Ge, Z. Yao, P. J. Brothers and X. Fu, J. Am. Chem. Soc., 2016, 138, 7705–7710 CrossRef CAS PubMed; (m) C. Hering-Junghans, P. Andreiuk, M. J. Ferguson, R. McDonald and E. Rivard, Angew. Chem., Int. Ed., 2017, 56, 6272–6275 ( Angew. Chem. , 2017 , 129 , 6368–6372 ) CrossRef CAS PubMed; (n) H. Jing, H. Ge, C. Li, Y. Jin, Z. Wang, C. Du, X. Fu and H. Fang, Organometallics, 2019, 38, 2412–2416 CrossRef CAS; (o) F. Diab, F. S. W. Aicher, C. P. Sindlinger, K. Eichele, H. Schubert and L. Wesemann, Chem. – Eur. J., 2019, 25, 4426–4434 CrossRef CAS PubMed.
- A. Hayatifar, A. Borrego, D. Bosek, M. Czarnecki, G. Derocher, A. Kuplicki, E. Lytle, J. Padilla, C. Paroly, G. Tubay, J. Vyletel and C. S. Weinert, Chem. Commun., 2019, 55, 10852–10855 RSC.
- G. Fegurson, J. F. Gallagher, D. Murphy, T. R. Spalding, C. Glidewell and H. D. Holden, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 1992, 48, 1228–1231 CrossRef.
- C. Glidewell and D. C. Liles, Acta Crystallogr., Sect. B: Struct. Crystallogr. Cryst. Chem., 1978, 34, 119–124 CrossRef.
- Z. Xie, R. Bau and C. A. Reed, J. Chem. Soc., Chem. Commun., 1994, 2519–2520 RSC.
- S. Popov, B. Shao, A. L. Bagdasarian, T. R. Benton, L. Zou, Z. Yang, K. N. Houk and H. M. Nelson, Science, 2018, 361, 381–387 CrossRef CAS PubMed.
- (a) G. Meißner, D. Dirican, C. Jäger, T. Braun and E. Kemnitz, Catal. Sci. Technol., 2017, 7, 3348–3354 RSC; (b) G. Meißner, K. Kretschmar, T. Braun and E. Kemnitz, Angew. Chem., Int. Ed., 2017, 56, 16338–16341 ( Angew. Chem. , 2017 , 129 , 16556–16559 ) CrossRef PubMed.
- (a) L. Maron, E. L. Werkema, L. Perrin, O. Eisenstein and R. A. Andersen, J. Am. Chem. Soc., 2005, 127, 279–292 CrossRef CAS PubMed; (b) A. R. Fout, J. Scott, D. L. Miller, B. C. Bailey, M. Pink and D. J. Mindiola, Organometallics, 2009, 28, 331–347 CrossRef CAS; (c) J. Chu, X. Han, C. E. Kefalidis, J. Zhou, L. Maron, X. Leng and Y. Chen, J. Am. Chem. Soc., 2014, 136, 10894–10897 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Full characterization, NMR spectra and X-ray data are provided. CCDC 1983059. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0cc01420f |
| This journal is © The Royal Society of Chemistry 2020 |