
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Unlocking the direct photocatalytic difluoromethylation of C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/h2_char_e001.gif) N bonds†
N bonds†
Alberto F.
Garrido-Castro
 a,
Andrea
Gini
a,
M. Carmen
Maestro
*a and
José
Alemán
*ab
a,
Andrea
Gini
a,
M. Carmen
Maestro
*a and
José
Alemán
*ab
aDepartment of Organic Chemistry, Universidad Autónoma de Madrid, 28049, Spain. E-mail: jose.aleman@uam.es; carmen.maestro@uam.es; Web: http://www.uam.es/jose.aleman
bInstitute for Advanced Research in Chemical Sciences (IAdChem), Universidad Autónoma de Madrid, 28049, Spain
First published on 27th February 2020
Abstract
The current study presents a direct CF2H radical addition to C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N bonds predicated on the photocatalytic activation of commercially available zinc difluoromethanesulfinate. The mild conditions in place lead to impressive structural diversity, as quinoxalinones and dibenzazepines, among others, are successfully functionalized.
N bonds predicated on the photocatalytic activation of commercially available zinc difluoromethanesulfinate. The mild conditions in place lead to impressive structural diversity, as quinoxalinones and dibenzazepines, among others, are successfully functionalized.
Fluorine stands as the most abundant halogen on Earth, yet it has played an insignificant role during the natural biosynthesis of organic molecules.1 Despite shortage of organofluorides in Nature, chemists have discovered and exploited the unique properties of fluorine-containing compounds for decades, flooding the field of pharmaceutical, agrochemical and material science with a wide toolbox of innovative and unique strategies to achieve fluorine incorporation.2 In the realm of drug discovery and development, the installation of fluoromethyl groups (–CFxHy) into organic molecules has received significant consideration.3 Fittingly, over 20% of the currently approved drugs contain one or more fluorine atoms in their scaffolds.4 Fluoroalkylated compounds generally display enhanced bioavailability and drug uptake given their: (i) higher lipophilicity than non-fluorinated analogues, leading to better membrane permeability, (ii) resistance towards oxidation, which results in increased metabolic stability, and (iii) improved binding selectivity.5 Pointedly, the difluoromethyl group (–CF2H) can serve as a suitable isostere to traditional hydrogen-bond donors such as alcohols, thiols or hydroxamic acid.6
Although trifluoromethylation processes have been studied extensively over the years,7 direct difluoromethylations have remained elusive.8 Insertion of the CF2H functionality into a specific target typically relies on multi-step methodologies, in which a CF2-FG derivative is attached to the desired site and then, the functional group (FG) is removed to generate the CF2H fragment.9 This shortcoming is clearly exemplified when reviewing CN bond difluoromethyl additions (Scheme 1A). Hu and co-workers have been at the forefront of this synthetic challenge, generating highly nucleophilic sulfonyl- and thio-difluoromethyl anions to achieve aldimine difluoromethylation, requiring initial activation and final sulfur removal (top, Scheme 1A).10 In this regard, the Hu group achieved a variant of this process through challenging activation of rather inert TMSCF2H.11 Prakash et al. have also reported an interesting follow-up on their trifluoromethylation strategy,12 in which addition of the Ruppert–Prakash reagent (TMSCF3), increased fluoride loading and subsequent reduction afforded the corresponding difluoromethylated amines (bottom, Scheme 1A).13 To the best of our knowledge, these two-electron approaches represent the only existing pathways to achieve difluoromethyl addition at the CN bond. Furthermore, the strong base, toxic reagents and restrictive experimental conditions limit range and applicability. Therefore, it would be highly desirable to develop a direct and benign CF2H addition to CN bonds.
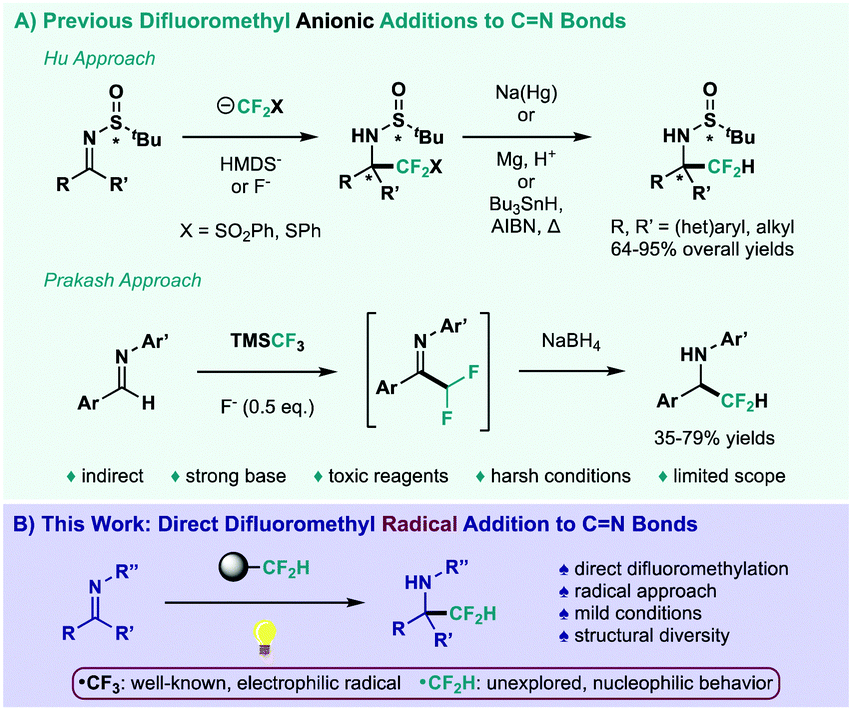 | ||
| Scheme 1 (A) Previous difluoromethyl anionic additions to CN bonds. (B) This work: direct difluoromethyl radical addition to CN bonds. | ||
Alternatively, photoredox catalysis has been established as a powerful tool for radical generation under milder reaction setups, while also proving to be extremely chemoselective regarding substrates outside their range of oxidative and reductive potential.14 In this context, radical fluoroalkylation has benefitted greatly from the recent renaissance in photochemistry,15 especially trifluoromethylation protocols.16 Nevertheless, the trifluoromethyl radical represents a markedly electrophilic species, impeding its addition onto an innately electrophilic CN bond. Notably, a remarkable regiochemical comparison between the CF3 and CF2H radicals was reported, achieving exclusive C–H functionalization at nucleophilic and electrophilic sites of heteroarenes, respectively.17 The use of the CF2H radical as a nucleophilic species, however, remains deeply unexplored.18 In fact, the direct photocatalytic difluoromethyl addition to CN bonds has never been accomplished to the best of our knowledge. Given the challenge this combination represents, we herein report the direct difluoromethylation of imines and its application to a wide array of CN bond-centric structures (Scheme 1B).
Difluoromethylation studies began with judicious selection of the reacting partners. Diphenyl-substituted aldimine 1a was chosen as model substrate because of its straightforward backbone, and methodical variations of its structure could give valuable information on its reactivity. Moreover, zinc difluoromethanesulfinate (DFMS) was quickly identified as an optimal CF2H source since it is commercially available, air-stable and easy to handle (see ESI†).17,19 Most importantly, it features a mild oxidation potential (Eox = +1.35 V vs. SCE in MeCN, see ESI† for voltammetry), thus possibly engaging in SET (Single-Electron Transfer) events with a large number of readily accessible photosensitizers (both organometallic complexes and organic dyes).14 A summary of the most noteworthy results during initial experimentation is shown in Table 1 (see ESI† for detailed optimization studies). Preliminary testing in the presence of standard Ir- and Ru-based photocatalysts (entries 1 and 2) yielded promising results. Following thorough photocatalyst screening, inexpensive rhodamine 6G (Rh-6G, entry 3) provided the best results when irradiated near its local absorbance maximum (540 nm).14 This xanthene-based dye, however, has displayed enhanced photocatalytic activity upon irradiation with a more energetic wavelength (450 nm).14 Gratifyingly, this behavior could be exploited, delivering a significant increase in yield of 2a (entry 4). Optimal conditions were reached after solvent screening and adjustment of DFMS loading (entries 5–7). Meanwhile, control experiments revealed that photocatalyst and light irradiation are indispensable for the reaction to take place (entries 8 and 9).
|
|
|||||
|---|---|---|---|---|---|
| Entrya | Photocatalyst | hν (nm) |
1a![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :DFMS molar ratio :DFMS molar ratio |
Solvent | Yieldc (%) |
| a Reaction conditions: 0.1 mmol scale using 1.0 equiv. of aldimine 1a and 2 mol% of photocatalyst in 1.0 mL of solvent during 16 h. b 360 mW single LED. See ESI for detailed experimental setup. c Determined by 1H NMR using MeNO2 as internal standard. d Isolated yield after flash chromatography. | |||||
| 1 | [Ir{dF(CF3)ppy}2(dtbbpy)]PF6 | 420 | 1:2 |
DMF | 35 |
| 2 | [Ru(bpy)3]Cl2·6H2O | 450 | 1:2 |
DMF | 33 |
| 3 | Rh-6G | 540 | 1:2 |
DMF | 42 |
| 4 | Rh-6G | 450 | 1:2 |
DMF | 54 |
| 5 | Rh-6G | 450 | 1:2 |
MeCN | 65 |
| 6 | Rh-6G | 450 | 1:1.5 |
MeCN | 76 (65)d |
| 7 | Rh-6G | 450 |
1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/b_char_2009.gif) :1 :1
|
MeCN | 77 (72) |
| 8 | — | 450 | 1:1 |
MeCN | 0 |
| 9 | Rh-6G | — | 1:1 |
MeCN | 0 |
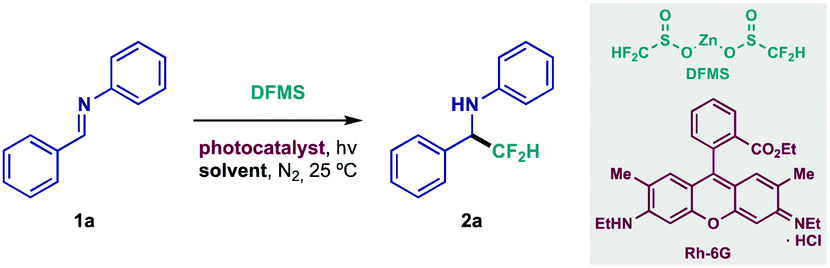
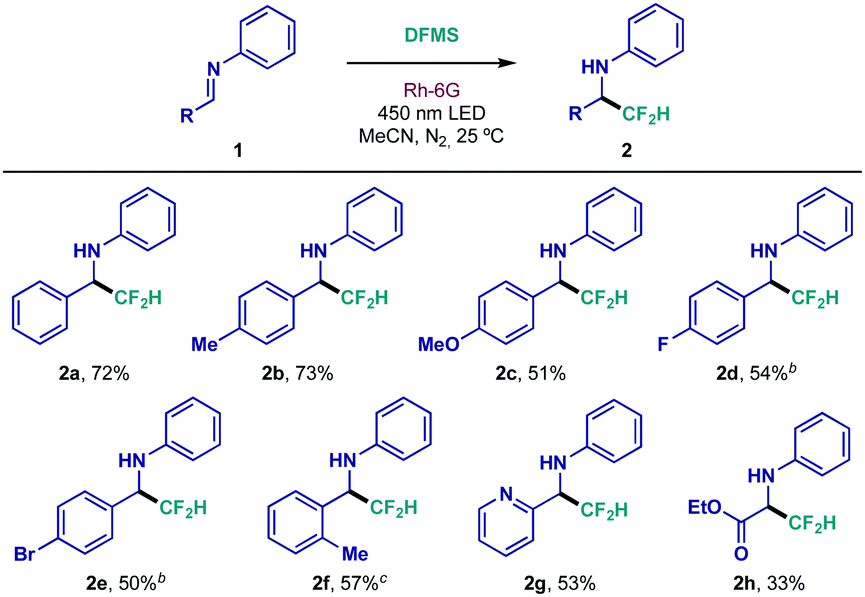
With the optimized reaction conditions in hand, the structural scope of this class of diarylimines was evaluated (Table 2). Electron-donating substitution on the benzaldehyde ring—p-Me and p-OMe—was well tolerated (2b and 2c, respectively). Halide compatibility was also achieved; the reaction proceeded smoothly in the presence of electron-withdrawing p-fluoride (2d), and the p-bromide functionality remained intact during preparation of difluoromethylated amine 2e. Sterically demanding o-substitution was also responsive to this protocol (2f). Remarkably, pyridyl-substituted imine 1g underwent chemoselective CF2H radical addition, providing the fluorinated heteroaryl amine with moderate yield (2g). Lastly, glyoxylate-derived imine 1h was successfully subjected to the newly developed conditions (2h). Even at its early developmental stage, this method had already showcased interesting applicability, giving access to 2e, which could be useful in orthogonal cross-coupling reactions, and unlocking difluoromethylated pyridyl and amino acid derivatives (2g and 2h, respectively). Further modifications were evaluated, yet N-phenyl substitution resulted essential for efficient reactivity, most likely due to the stabilizing effect it has on the N-centered radical intermediate that is formed upon CF2H radical addition to the CN bond, and the intrinsic stability of the imine under the reaction conditions.
Following this initial scope evaluation, exploration of relevant cores featuring CN bonds susceptible to CF2H radical addition was taken into account.17 Fused nitrogen heterocycles such as quinolines, quinoxalinones and dibenzazepines rapidly surfaced as interesting substrates given their privileged position among bioactive scaffolds.20 Particularly, quinolines presented an intriguing case due to their multiple reactive sites. The set of results shown in Scheme 2 highlights the importance of the generated intermediates in this process. Unsubstituted quinoline 3a displayed low reactivity under the photocatalytic conditions, giving rise to the 1,4-product (4a) as major regioisomer – only trace amount of 1,2-adduct was detected, while the unreacted starting material could be recovered unaltered. Predictably, tailoring of the heterocyclic framework could lead to 1,4- or 1,2-functionalization (4b and 4c, respectively) in considerably higher yields. The acridine moiety 3d, however, afforded the dearomatized 1,4-difluoromethylated adduct 4d; a key observation underlining the inability of the acridine-derived N-centered radical intermediate to rearomatize since it would require unfavorable dearomatization of its aromatic rings in the first place.
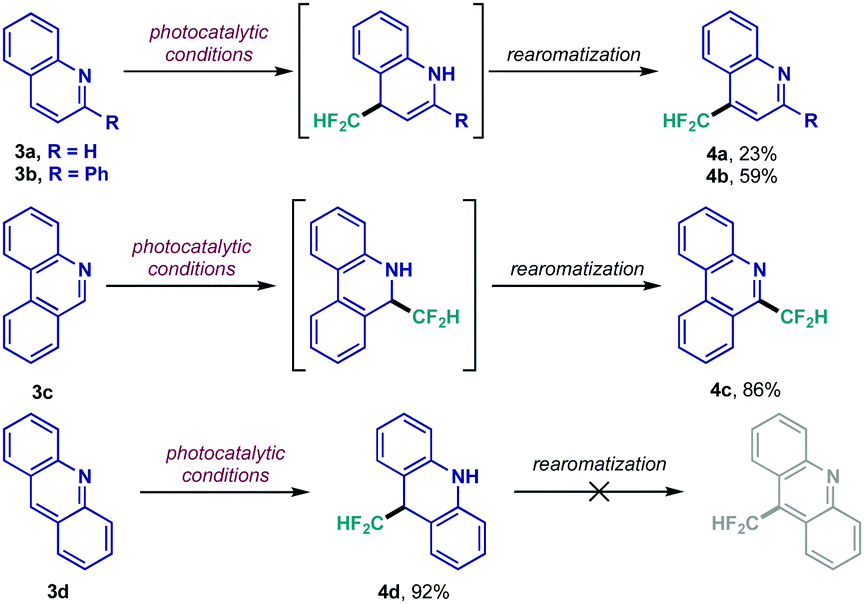 | ||
| Scheme 2 Exploration of the photocatalytic CF2H radical addition to quinolines 3. | ||
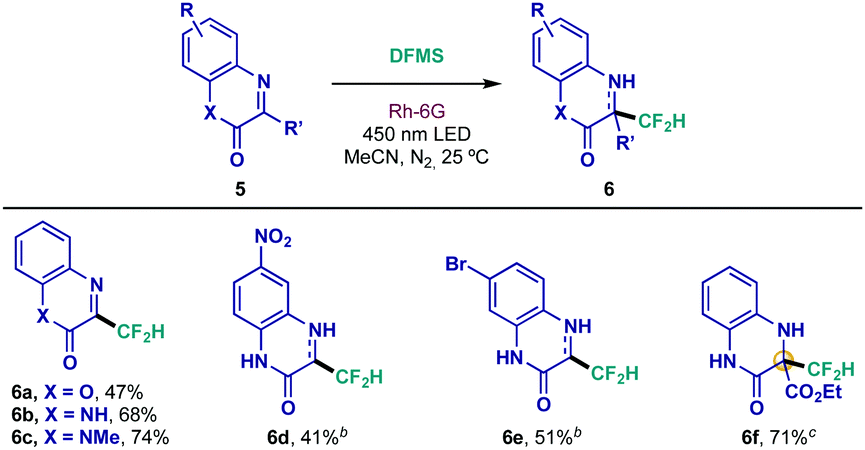
Additionally, quinoxalinones have shown an impressive pharmacological profile, displaying antimicrobial, antiviral and antitumor activities, among others.21 Therefore, application of this new difluoromethylation protocol could lead to interesting substrates given the aforementioned properties of the CF2H group. As depicted in Table 3, 4-azacoumarin, quinoxalinone and N-methyl quinoxalinone underwent smooth difluoromethylation under the optimized conditions (6a–6c). Interestingly, inclusion of a typically restrictive nitro group in the structure was well tolerated (6d), whereas brominated scaffold 6e was prepared in adequate yield. Most impressively, quaternary difluoromethylated adduct 6f was achieved with the highly interesting iminoester derivative 5f, an unprecedented result in the field of direct difluoromethylation of imines.
|
a Reaction conditions: 0.1 mmol scale using 1.0 equiv. of heterocycle 5, 0.075 mmol of DFMS and 2 mol% of Rh-6G in 1.0 mL of MeCN during 16 h. Isolated yields are indicated under each entry.
b
6d and 6e were obtained as a mixture of aromatized and dearomatized products (39:61 and 35:65, respectively; ratio determined by 1H NMR).
c Reaction performed with 0.1 mmol of DFMS during 48 h.
|
|---|
|
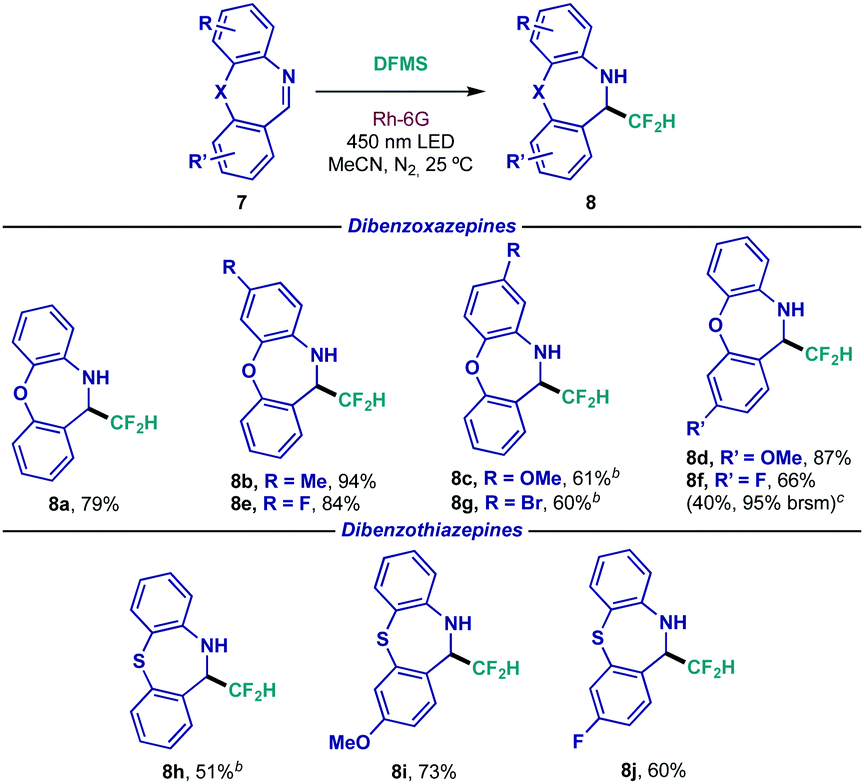
| a Reaction conditions: 0.1 mmol scale using 1.0 equiv. of dibenzazepine 7, 0.1 mmol of DFMS and 2 mol% of Rh-6G in 1.0 mL of MeCN during 48 h. Isolated yields are indicated under each entry. b Reaction time: 16 h. c Reaction performed on a 0.75 mmol scale. brsm = based on recovered starting material. |
|---|
|
Among pharmaceutical motifs, dibenzazepines constitute an essential component of second generation or atypical antipsychotics.22 As such, these tricyclic moieties were subjected to the difluoromethylating conditions (Table 4). Evaluation of the structural scope rendered exciting results as dibenzoxazepine 7a underwent photocatalytic difluoromethylation efficiently (8a). Electron-donating (8b–8d) and electron-withdrawing (8e and 8f) bias was once again tolerated under the present conditions, regardless of the placement of the substitution. As observed in previous instances, amine 8g bearing the easily cross-coupled bromide functionality could be prepared in good yield. In an attempt to scale up the reaction, substrate 8f revealed modest results at a 0.75 mmol scale. As for analogous dibenzothiazepines, performance appeared to feature similar reactivity to their oxo-analogues, giving access to unbiased (8h), electron-rich (8i) and electron-poor (8j) substrates in synthetically useful yields.
From a mechanistic standpoint, a proposal based on a series of experimental trials is outlined in Scheme 3 (see ESI† for detailed mechanistic studies). Initial excitation of the photocatalyst Rh-6G under visible light irradiation leads to the formation of the excited species *Rh-6G. Stern–Volmer quenching studies indicate that this species is quenched by the difluoromethylating reagent DFMS affording ˙CF2H through single-electron oxidation and extrusion of SO2 (Eox = +1.35 V vs. SCE). The resulting difluoromethyl radical reacts with the CN bond acting as a pseudo-nucleophile en route to aminyl radical intermediate I. At this stage, two possible outcomes could be expected: (i) a Hydrogen Atom Transfer (HAT) event with intermediate I, generating the final difluoromethylated product 2a; or (ii) a single-electron reduction of intermediate I, followed by proton abstraction to yield the final adduct 2a. Throughout the development of this protocol, several H atom donors were tested, observing no positive effect on the final yield. In fact, incremental addition of redox-inactive 1,4-cyclohexadiene led to decreased yield, or even total inhibition of the reaction when used as co-solvent (bottom right, Scheme 3). Consequently, the most plausible pathway for intermediate I would involve reduction to form intermediate II. This reduction step could be enforced by Rh-6G˙− – closing the photocatalytic cycle – or could initiate a radical chain mechanism by oxidation of DFMS (bottom left, Scheme 3). However, quantum yield determination (ϕ < 1) suggests that the catalytic reduction is taking place, although a radical chain process cannot be ruled out.23 Lastly, use of deuterated solvent (CD3CN) confirmed acetonitrile as the proton source for the final step of the reaction, as evidenced by the formation of the fully N-deuterated product.
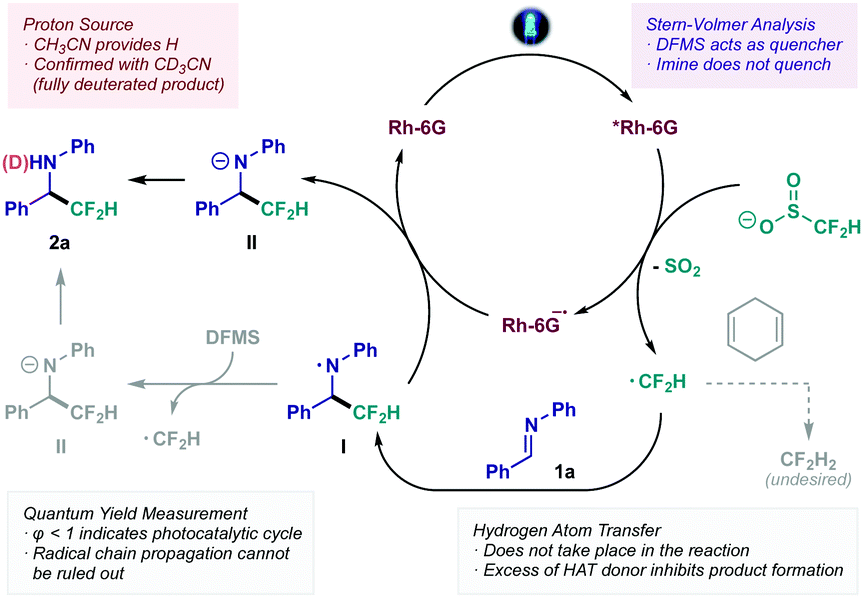 | ||
| Scheme 3 Mechanistic proposal for the photocatalytic CF2H radical addition to CN bonds. | ||
In conclusion, a direct difluoromethyl addition to CN bonds has been developed. The photocatalytic activation of commercially available DFMS as difluoromethylating reagent delivers a new radical approach which benefits from the mild conditions to achieve impressive structural diversity; diaryl-substituted aldimines, quinolines, quinoxalinones and dibenzazepines are successfully functionalized with the pharmacologically crucial CF2H group. Finally, a mechanistic proposal based on several experimental trials is presented, in which ˙CF2H addition and subsequent reduction of the aminyl radical constitute the key steps of the process.
Financial support was provided by the European Research Council (ERC-CoG, contract number: 647550), Spanish Government (RTI2018-095038-B-I00), “Comunidad de Madrid” and European Structural Funds (S2018/NMT-4367).
Conflicts of interest
There are no conflicts to declare.Notes and references
- (a) D. B. Harper and D. O’Hagan, Nat. Prod. Rep., 1994, 11, 123 RSC; (b) X.-H. Xu, G.-M. Yao, Y.-M. Li, J.-H. Lu, C.-J. Lin, X. Wang and C.-H. Kong, J. Nat. Prod., 2003, 66, 285 CrossRef CAS PubMed.
- (a) C. Hollingworth and V. Gouverneur, Chem. Commun., 2012, 48, 2929 RSC; (b) T. Liang, C. N. Neumann and T. Ritter, Angew. Chem., Int. Ed., 2013, 52, 8214 CrossRef CAS PubMed.
- K. L. Kirk, Org. Process Res. Dev., 2008, 12, 305 CrossRef CAS.
- (a) K. Müller, C. Faeh and F. Diederich, Science, 2007, 317, 1881 CrossRef PubMed; (b) J. Wang, M. Sánchez-Roselló, J. L. Aceña, C. del Pozo, A. E. Sorochinsky, S. Fustero, V. A. Soloshonok and H. Liu, Chem. Rev., 2014, 114, 2432 CrossRef CAS PubMed.
- (a) S. Purser, P. R. Moore, S. Swallow and V. Gouverneur, Chem. Soc. Rev., 2008, 37, 320 RSC; (b) W. K. Hagmann, J. Med. Chem., 2008, 51, 4359 CrossRef CAS PubMed.
- (a) N. A. Meanwell, J. Med. Chem., 2011, 54, 2529 CrossRef CAS PubMed; (b) Y. Zafrani, D. Yeffet, G. Sod-Moriah, A. Berliner, D. Amir, D. Marciano, E. Gershonov and S. Saphier, J. Med. Chem., 2017, 60, 797 CrossRef CAS PubMed.
- (a) J.-A. Ma and D. Cahard, J. Fluorine Chem., 2007, 128, 975 CrossRef CAS; (b) O. A. Tomashenko and V. V. Grushin, Chem. Rev., 2011, 111, 4475 CrossRef CAS PubMed; (c) A. Studer, Angew. Chem., Int. Ed., 2012, 51, 8950 CrossRef CAS PubMed.
- (a) J. Hu, W. Zhang and F. Wang, Chem. Commun., 2009, 7465 RSC; (b) D. E. Yerien, S. Barata-Vallejo and A. Postigo, Chem. – Eur. J., 2017, 23, 14676 CrossRef CAS PubMed.
- C. Ni, M. Hu and J. Hu, Chem. Rev., 2015, 115, 765 CrossRef CAS PubMed.
- (a) Y. Li and J. Hu, Angew. Chem., Int. Ed., 2005, 44, 5882 CrossRef CAS PubMed; (b) Y. Li and J. Hu, Angew. Chem., Int. Ed., 2007, 46, 2489 CrossRef CAS PubMed; (c) J. Liu, Y. Li and J. Hu, J. Org. Chem., 2007, 72, 3119 CrossRef CAS PubMed; (d) J. Liu and J. Hu, Chem. – Eur. J., 2010, 16, 11443 CrossRef CAS PubMed; (e) S. Fustero, J. Moscardó, M. Sánchez-Roselló, E. Rodríguez and P. Barrio, Org. Lett., 2010, 12, 5494 CrossRef CAS PubMed . Via Reformatsky reactions, see: ; (f) T. Poisson, M.-C. Belhomme and X. Pannecoucke, J. Org. Chem., 2012, 77, 277 CrossRef PubMed; (g) C.-R. Cao, M. Jiang and J.-T. Liu, Eur. J. Org. Chem., 2015, 1144 CrossRef CAS.
- Y. Zhao, W. Huang, J. Zheng and J. Hu, Org. Lett., 2011, 13, 5342 CrossRef CAS PubMed.
- G. K. S. Prakash, M. Mandal and G. A. Olah, Angew. Chem., Int. Ed., 2001, 40, 589 CrossRef CAS PubMed.
- G. K. S. Prakash, R. Mogi and G. A. Olah, Org. Lett., 2006, 8, 3589 CrossRef CAS PubMed.
- Visible Light Photocatalysis in Organic Chemistry, ed. C. R. J. Stephenson, T. P. Yoon and D. W. C. MacMillan, Wiley-VCH, Weinheim, 2018 Search PubMed.
- (a) T. Chatterjee, N. Iqbal, Y. You and E. J. Cho, Acc. Chem. Res., 2016, 49, 2284 CrossRef CAS PubMed; (b) S. Barata-Vallejo, M. V. Cooke and A. Postigo, ACS Catal., 2018, 8, 7287 CrossRef CAS; (c) T. Koike and M. Akita, Org. Biomol. Chem., 2019, 17, 5413 RSC.
- T. Koike and M. Akita, Acc. Chem. Res., 2016, 49, 1937 CrossRef CAS PubMed.
- Y. Fujiwara, J. A. Dixon, R. A. Rodriguez, R. D. Baxter, D. D. Dixon, M. R. Collins, D. G. Blackmond and P. S. Baran, J. Am. Chem. Soc., 2012, 134, 1494 CrossRef CAS PubMed.
- (a) J. Rong, C. Ni and J. Hu, Asian J. Org. Chem., 2017, 6, 139 CrossRef CAS; (b) A. Lemos, C. Lemaire and A. Luxen, Adv. Synth. Catal., 2019, 361, 1500 CrossRef CAS PubMed.
- (a) Y. Fujiwara, J. A. Dixon, F. O’Hara, E. D. Funder, D. D. Dixon, R. A. Rodriguez, R. D. Baxter, B. Herlé, N. Sach, M. R. Collins, Y. Ishihara and P. S. Baran, Nature, 2012, 492, 95 CrossRef CAS PubMed; (b) F. O’Hara, R. D. Baxter, A. G. O’Brien, M. R. Collins, J. A. Dixon, Y. Fujiwara, Y. Ishihara and P. S. Baran, Nat. Protoc., 2013, 8, 1042 CrossRef PubMed.
- Heterocycles in Life and Society: An Introduction to Heterocyclic Chemistry, Biochemistry and Applications, ed. A. F. Pozharskii, A. T. Soldatenkov and A. R. Katritzky, Wiley, Chichester, 2011, pp. 139–183 Search PubMed.
- L. Shi, W. Hu, J. Wu, H. Zhou, H. Zhou and X. Li, Mini-Rev. Med. Chem., 2018, 18, 392 CrossRef CAS PubMed.
- S. Jafari, F. Fernandez-Enright and X.-F. Huang, J. Neurochem., 2012, 120, 371 CrossRef CAS PubMed.
- Indeed, a radical chain reaction hypothesis might be excluded when ϕ < 1, although quantum yield determination can only serve as conclusive evidence when ϕ > 1; see: M. A. Cismesia and T. P. Yoon, Chem. Sci., 2015, 6, 5426 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0cc01353f |
| This journal is © The Royal Society of Chemistry 2020 |