
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Controlling the properties of the micellar and gel phase by varying the counterion in functionalised-dipeptide systems†
Kate
McAulay
a,
Pedro Agís
Ucha
ab,
Han
Wang
c,
Ana M.
Fuentes-Caparrós
a,
Lisa
Thomson
 a,
Osama
Maklad
d,
Nikul
Khunti
e,
Nathan
Cowieson
e,
Matthew
Wallace
f,
Honggang
Cui
c,
Robert J.
Poole
d,
Annela
Seddon
gh and
Dave J.
Adams
*a
a,
Osama
Maklad
d,
Nikul
Khunti
e,
Nathan
Cowieson
e,
Matthew
Wallace
f,
Honggang
Cui
c,
Robert J.
Poole
d,
Annela
Seddon
gh and
Dave J.
Adams
*a
aSchool of Chemistry, University of Glasgow, Glasgow, G12 8QQ, UK. E-mail: dave.adams@glasgow.ac.uk
bDepartment of Chemical Engineering, Faculty of Sciences, 18071 Granada, Spain
cDepartment of Chemical and Biomolecular Engineering, Whiting School of Engineering, Johns Hopkins University, 3400 North Charles Street, Baltimore, MD 21218, USA
dSchool of Engineering, University of Liverpool, Liverpool, L69 3GH, UK
eDiamond Light Source Ltd, Harwell Science and Innovation Campus, Didcot, OX11 0QX, UK
fSchool of Pharmacy, University of East Anglia, Norwich Research Park, Norwich, NR4 7TJ, UK
gSchool of Physics, HH Wills Physics Laboratory, University of Bristol, Tyndall Avenue, Bristol, BS8 1TL, UK
hBristol Centre for Functional Nanomaterials, HH Wills Physics Laboratory, University of Bristol, Tyndall Avenue, Bristol, BS8 1TL, UK
First published on 5th March 2020
Abstract
The micellar aggregates formed at high pH for dipeptide-based gelators can be varied by using different alkali metal salts to prepare the solutions. The nature of the micellar aggregates directly affects the properties of the resulting gels.
Gels are formed by low molecular weight gelators by their self-assembly into long anisotropic structures.1–3 These entangle to form the matrix. The gel properties are controlled by the mechanical properties of the fibres and how these entangle, cross-link and otherwise form junctions.4 In terms of designing gels, there are currently two key problems.5 First, it is difficult to predict in advance whether a specific molecule will be a gelator or not, with very similar chemical structures having very different gelation efficiency for example.5 Computational prediction tools are improving, but are still limited in scope.6–8 Second, it is not possible to predict the properties of the gels if they are formed. These two issues hold the field back and many useful systems are discovered accidentally.5,9
A more pragmatic approach is to find a molecule that is an effective gelator and then vary the assembly process such that gels with required properties are formed.3 The gel properties can be adjusted by varying the gelation method or by changing the composition or conditions slightly.3,10 For hydrogels, the situation is complicated by potential pre-aggregation prior to gelation. Many hydrogelators are relatively hydrophobic (to drive poor solubility and gelation) and so the pre-gelled, “soluble” state can contain micellar aggregates.11,12 Hence, on applying the gelation trigger, there is a transition from micellar aggregate to gel state. In the case of functionalised dipeptide-based gelators, the micellar state can template the structures formed in the gel state.11 For a single example, the functionalised dipeptide 2NapFF (Fig. 1a), the molecules pack into hollow tubes in the micellar state at high pH.11 When the system is gelled, the hollow core is lost, and lateral association leads to elliptical structures that entangle to form the gel.13
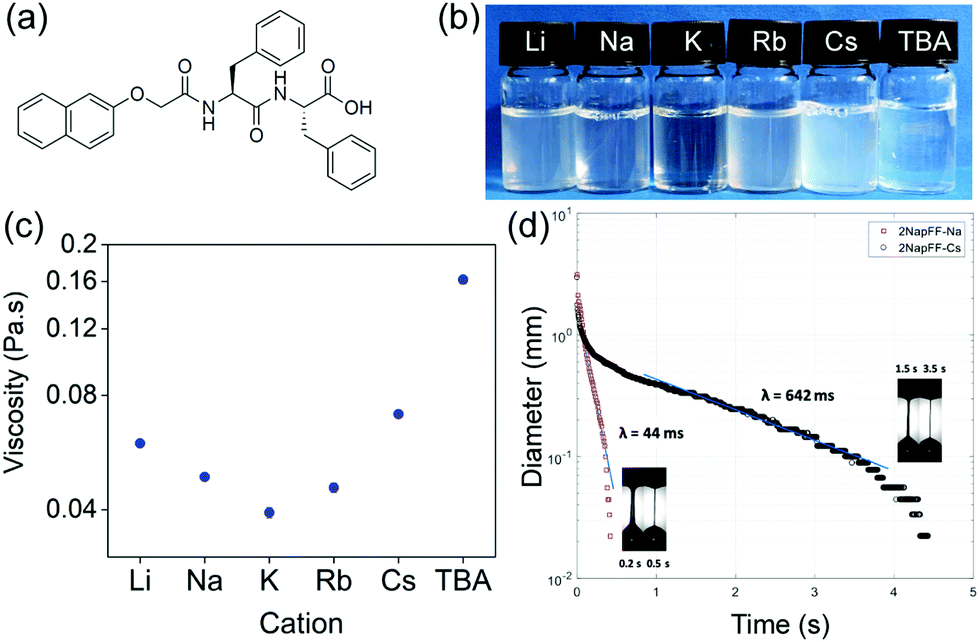 | ||
| Fig. 1 (a) Structure of 2NapFF; (b) solutions of 2NapFF at pH 11 at 10 mg mL−1, cation displayed on lid; (c) comparison of shear viscosity values at 10 s−1 for solutions prepared at 10 mg mL−1 using different salts; (d) Variation of filament diameter versus time from CaBER experiment for Na-2NapFF and Cs-2NapFF highlighting significant differences caused by choice of salt. Note “time” shifted such that region where filament enters capillary thinning regime can be plotted on same axes for both results: Insets in each subfigure show high-speed camera images of filament shapes which enabled us to determine where to apply exponential fit and calculate relaxation time (λ). | ||
We can therefore consider functionalised dipeptides such as 2NapFF as surfactants at high pH.13 It should therefore be possible to vary the micellar aggregates formed from a single gelator by changing parameters such as the nature of the counterion. From conventional surfactant theory, this should lead to a change in the packing parameter,14 and perhaps a change in the micellar aggregates formed.15,16 Varying the micellar aggregate should allow us to control the apparent pKa of the aggregate,17,18 and hence the pH at which gels form, and likely the properties of gels that are formed.
To test this hypothesis, we prepared solutions of 2NapFF at a pH of 11 using one of a number of hydroxide salts: LiOH, NaOH, KOH, RbOH, CsOH or tetrabutylammonium hydroxide (TBAOH) to deprotonate the terminal carboxylic acid. At a concentration of 10 mg mL−1, viscous solutions were prepared with all salts, with some change in the degree of transparency/turbidity (Fig. 1b). As expected from our previous work,19 at 10 mg mL−1, the solutions exhibit viscoelastic shear thinning behaviour (Fig. S1, ESI†). The viscosity of the solutions depends directly on the salt used to make the initial gelator solution. Solutions formed using TBAOH were the most viscous, whilst those formed with KOH were the least viscous at a shear rate of 10 s−1 (Fig. 1c), although we note that the changes in shear viscosity are quite modest and within repeatability apart from that for the TBAOH.
Each of the systems also exhibits viscoelastic behaviour and significant extensional viscosity in a capillary breakup extensional rheometer (CaBER), with the relaxation time measured being dependent on the salt used. Here, an unstable filament is formed with continuous decrease in diameter under the combined effect of surface tension and viscoelastic extensional forces until it finally breaks. The high-speed camera images of the filament enabled us to determine where the CaBER theory can be applied and consequently apply the exponential fit to determine the relaxation time.20 Representative plots are shown in Fig. 1d. We have previously shown that the extensional viscosity increases dramatically for Na-2NapFF after a heat–cool cycle.21 Here, the effect of the heat–cool cycle can be seen to increase the characteristic relaxation time (λ) for Na-2NapFF by one order of magnitude as shown previously.21
Depending on the salt used, it is possible to either have a large relaxation time without recourse to a heat–cool cycle (Cs-2NapFF), have a low relaxation time prior to a heat–cool cycle which leads to a significant increase (Na-2NapFF or Rb-2NapFF) or have a low relaxation time either both or after the heat–cool cycle (TBA-2NapFF) (Table 1). Overall, the choice of salt or use of a heat–cool cycle can significantly modify the extensional properties of the solutions from essentially “Newtonian-like” with low relaxation time (<0.05 s) to highly “stringy” elastic fluids with very large relaxation times. This behaviour is in marked contrast to the shear viscosity where the changes are more modest.
| Relaxation time | Li | Na | K | Rb | Cs | TBA |
|---|---|---|---|---|---|---|
| Before H–C (λB) (ms) | 295 | 44 | 517 | 104 | 642 | 36 |
| After H–C (λA) (ms) | 483 | 343 | 827 | 2350 | 1760 | 41 |
| Ratio λA/λB | 1.6 | 7.8 | 1.6 | 22.6 | 2.7 | 1.1 |
All of these differences imply that there are likely different structures formed in these systems. To probe this, we used cryo-TEM and small angle X-ray scattering (SAXS). Note that drying effects are common and also crystal structures often do not represent the solvated state in these systems.3 The cryo-TEM images show that there are differences in structure. Li-2NapFF shows the presence of long, helical structures (Fig. 2a). Na-2NapFF, Rb-2NapFF and Cs-2NapFF show the presence of long, uniform structures (example data shown for Rb-2NapFF in Fig. 2b). K-2NapFF shows similar long, uniform structures, as well as the presence of some ribbons. TBA-2NapFF shows the presence of long, uniform structures, with a significantly smaller diameter than the Na-, Rb-, and Cs-2NapFF (Fig. 2c). Cryo-TEM data for all samples are shown in Fig. S2 (ESI†). Cryo-TEM can only provide a snapshot of a small number of the structures present. Hence, to confirm the cryo-TEM data, we used small angle X-ray scattering (SAXS) as a bulk technique that can probe the samples.
 | ||
| Fig. 2 Cryo-TEM of (a) Li-2NapFF; (b) Rb-2NapFF; (c) TBA-2NapFF. The scale bar represents 100 nm in each case. (d) SAXS data for all samples, with the data shown as open symbols and the fit as a red line. Note the data are offset on the intensity scale for ease of discrimination. | ||
The SAXS data also show clearly that there are differences between the samples (Fig. 2d and Table S1, ESI†). To fit Li-2NapFF, we were guided by the TEM data, which shows the presence of helical structures; thus, we fitted the SAXS data at high pH for Li-2NapFF to a flexible elliptical cylinder as demonstrated elsewhere.22 The minor axis radius is found to be 3.1 nm and the major axis radius is 6.2 nm. SAXS data for Na-2NapFF, K-2NapFF, Cs-2NapFF and Rb-2NapFF were best fitted to a flexible cylinder as we have shown for Na-2NapFF.21 The radii for Na-2NapFF, K-2NapFF and Rb-2NapFF were similar (4.1 nm, 4.3 nm and 4.0 nm respectively), and commensurate with previous data for Na-2NapFF.21 Cs-2NapFF has a radius slightly larger than for the other examples (4.6 nm), however a Schulz polydispersity term had to be included into the fit for Rb-2NapFF and Cs-2NapFF, reflecting that there are a range of structures present in the solutions. TBA-2NapFF could be fitted to a flexible cylinder with a power law term. The small radius of 1.4 nm found by SAXS reflects the smaller size measured by TEM. Full details of the fits and fitted parameters can be found in the ESI† (Table S1).
In all systems, a significant interaction of the cation with the 2NapFF structures is detected (Fig. 3). The 7Li, 23Na and 133Cs resonances exhibit quadrupolar splitting while the 87Rb resonance is significantly broadened relative to the native hydroxide (Fig. S4, ESI†). The 39K resonance in K-2NapFF is broadened beyond detection. The 1H resonances of TBA are severely broadened so that no 1H–1H coupling along the alkyl chain can be detected. D2O and dioxane-d8 were used as complementary 2H probes.23 The splitting of these resonances depends upon both the degree of alignment and the solvation of the structures. With the exception of the TBA-2NapFF, the splitting of all or either of the D2O, dioxane-d8 or cation resonances indicates alignment of the structures. The high viscosity of TBA-2NapFF may prevent alignment. The relative splitting of the 2H resonances of D2O and dioxane-d8 is different in all samples, indicating that the solvation of the structures is different in each case.
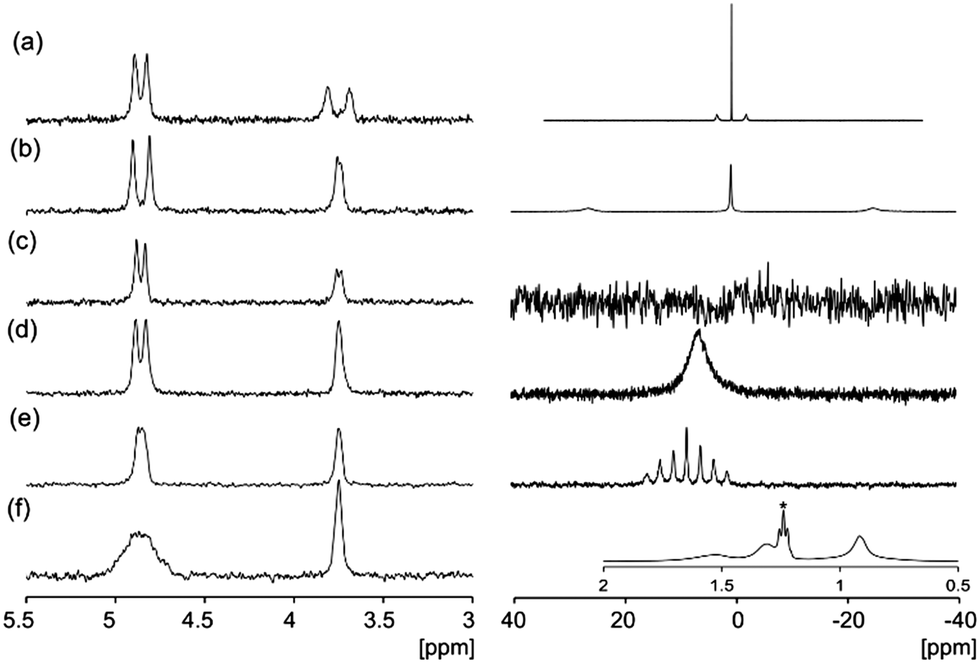 | ||
| Fig. 3 2H (left) and cation (right) NMR: solutions at 10 mg mL−1 of (a) Li-2NapFF; (b) Na-2NapFF; (c) K-2NapFF; (d) Rb-2NapFF; (e) Cs-2NapFF; (f) TBA-2NapFF, 1H, inset, the peak marked * corresponds to 2 μL mL−1 ethanol added to provide a comparison with the TBA resonances. All spectra are referenced to 2H dioxane at 3.75 ppm.24 | ||
The apparent pKa of the terminal carboxylic acid is different in all cases, varying from 9.6 to 4.9 (titration data shown in Fig. S5, ESI†). In each case, there are two apparent pKa, despite the single ionisable group as we have described previously.13 The differences in apparent pKa directly translate into differences in the pH at which gels are formed. To induce gelation, we use the hydrolysis of glucono-D-lactone (GdL), which slowly forms gluconic acid.25 Adding a solution of the 2NapFF to GdL leads to a slow pH decrease and the formation of a gel in each case (Fig. 4a and Fig. S6, ESI†). The final pH after 18 hours in each case was 3.6. There are some differences in the turbidity of the final gels (Fig. 4a), with the gel formed from Rb-2NapFF giving a significantly more translucent gel compared to the rest. Following the gelation process with time using small amplitude oscillatory shear (SAOS) rheology shows clear differences between the systems (Fig. 4b–g). The Li-2NapFF, Na-2NapFF and K-2NapFF systems have fairly sharp transitions in the storage modulus (G′) and loss modulus (G′′) at 2 h, 0.5 h, and 1.3 h respectively. In comparison, Rb-2NapFF, Cs-2NapFF and TBA-2NapFF systems show more gradual transitions to the gel phase. This can be connected to the pKa, values; when the pKa is higher, the pH matching the pKa is reached more quickly. The hydrolysis is base catalysed,26 so there are likely limited effects on changing the cation.
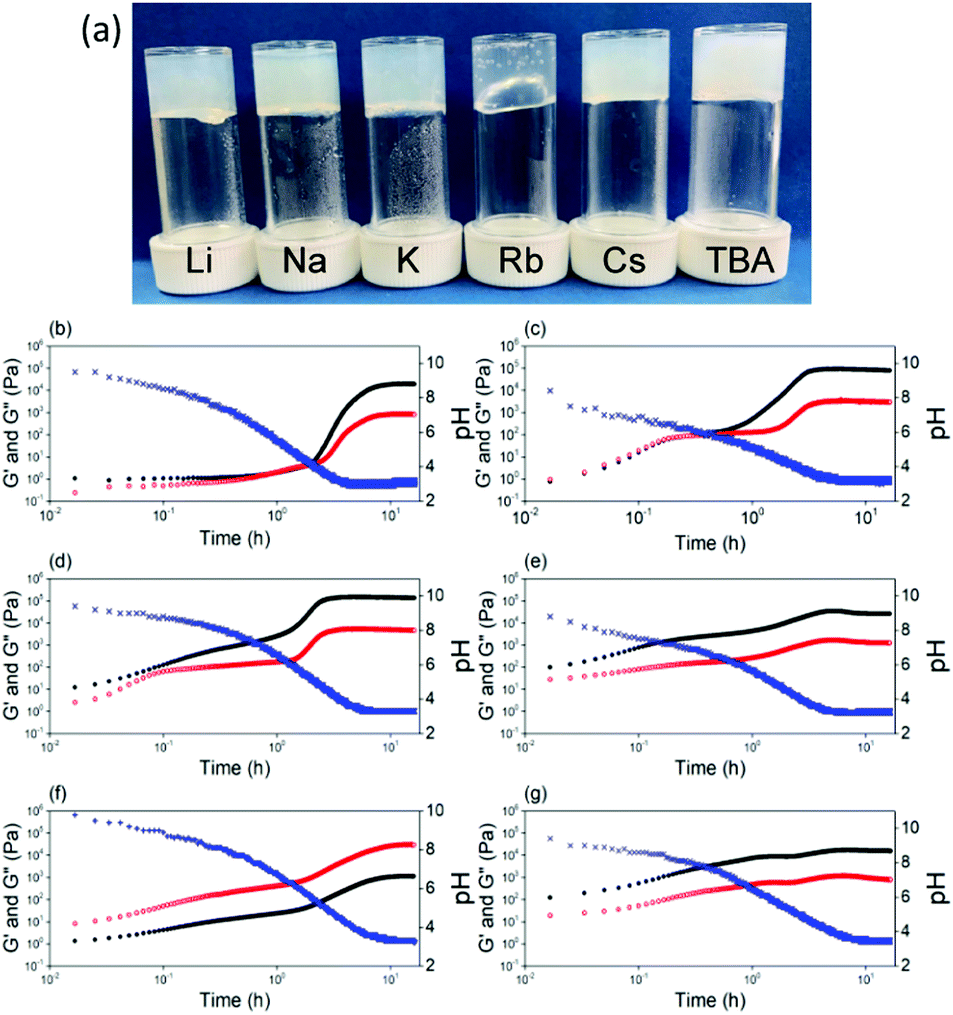 | ||
| Fig. 4 (a) Photographs of gels prepared at pH 3.6 at 10 mg mL−1, cation displayed on lid. Time sweep SAOS rheology showing the gel evolution on addition of GdL to solutions of (b) Li-2NapFF; (c) Na-2NapFF; (d) K-2NapFF; (e) Rb-2NapFF; (f) Cs-2NapFF; (g) TBA-2NapFF. The blue data is for pH, the black data for G′ and the red data for G′′. | ||
Being able to control the pH at which gels form opens up more opportunities. It is also important to be able to vary the stiffness of hydrogels. Gels with tuneable stiffnesses can be used to direct stem cell differentiation for example.27 In general, stiffness for such gels is varied by varying the concentration of gelator,1 which also tends to lead to other changes in properties. Here, we can vary the pH at which gelation occurs, the stiffness of the final gels, as well as the strain at which the gels break at a single concentration of gelator by varying the initial salt used. As we descend the periodic table, a general, but modest, increase in G′ is observed (Fig. S8, ESI†). In the case of the organic cation, TBA, the gel stiffness is significantly lower compared to using the inorganic salts (Fig. S8, ESI†). Descending the periodic table form Li to Cs results in the gels breaking at lower strains, showing that whilst there are only modest changes in the stiffness, the gels differ significantly in their absolute strength. For TBA-2NapFF gels, G′ never becomes less than G′′. Interestingly, the thermal behaviour is also different (Fig. S9, ESI†). For example, the Cs-2NapFF gel is stable to 90 °C, with little change in rheological properties before and after heating. The TBA-2NapFF gels melts at around 70 °C, and the rheological properties do not recover after heating; this may be due to the thinner structures present in TBA-2NapFF (see SAXS below and ESI†) having the least intermolecular interactions leading to a lower stability.
SAXS was performed on the gels formed from each high pH solution (it was found to be extremely difficult to probe the gels using cryo-TEM due to their stiffness). The scattering patterns and fits are shown in Fig. S3 (ESI†). In brief, the SAXS data for the gels show that the micellar aggregates formed at high pH directly link to the structures present in the gel phase, highlighting the importance in controlling the micellar structure.
Adding different salts to enzyme-triggered dipeptide gels has been reported by Ulijn's group.28 Differences were observed which were linked to the Hofmeister series; the differences in the resulting gel properties were ascribed to the salts affecting the enzyme network as opposed to the structures formed by the dipeptides.28 At high pH, the nature of the anion has been shown to be important and again can be linked to the Hofmeister series.29,30 For example, Roy et al. showed that the micellar aggregates formed from a Fmoc-dipeptide at pH 8 were fibres when sodium phosphate or sodium chloride were added, but spherical aggregates were formed when sodium thiocyante was added.29 In this case, gelation is driven by charge screening. Gelation for 2NapFF can be driven at high pH by adding salts to charge screen, or by addition of a divalent cation which cross-links the anisotropic structures present at high pH.30 However, our work here shows a different concept. Instead of gelation by charge screening, we are varying the aggregates formed by changing the packing in the micellar phase by the size of the cation. These different micellar aggregates template the structures formed in the gel phase when the pH is decreased. This directly leads to differences in the pH at which gels form as well as the mechanical properties for the gels.
In conclusion, it is possible to vary the micellar aggregates that are formed at high pH in solutions of a functionalised dipeptide by simply varying the salt used to raise the pH. The change in cation results in different packing in the micellar phase and the formation of different structures. The resulting solutions show different extensional viscosities, different behaviour on heating and cooling, and have different pKa values. As a result, gels are formed at different rates when the pH is decreased, and the final gels have different properties. This methodology opens up many opportunities with this class.
DA thanks the EPSRC for a Fellowship (EP/L021978/1), which also funded KM. AMFC and LT thank the University of Glasgow for funding. MW thanks The Royal Commission for the Exhibition of 1851 for a Research Fellowship. RP thanks the EPSRC for a Fellowship (EP/M025187/1). The Ganesha X-ray scattering apparatus was purchased under EPSRC Grant ‘Atoms to Applications’ (EP/K035746/1). This work benefitted from the SasView software, originally developed by the DANSE project under NSF award DMR-0520547.
Conflicts of interest
There are no conflicts to declare.Notes and references
- P. Terech and R. G. Weiss, Chem. Rev., 1997, 97, 3133–3160 CrossRef CAS PubMed.
- L. A. Estroff and A. D. Hamilton, Chem. Rev., 2004, 104, 1201–1218 CrossRef CAS PubMed.
- E. R. Draper and D. J. Adams, Chem, 2017, 3, 390–410 CAS.
- S. Sathaye, A. Mbi, C. Sonmez, Y. Chen, D. L. Blair, J. P. Schneider and D. J. Pochan, Wiley Interdiscip. Rev.: Nanomed. Nanobiotechnol., 2015, 7, 34–68 CAS.
- R. G. Weiss, J. Am. Chem. Soc., 2014, 136, 7519–7530 CrossRef CAS PubMed.
- J. K. Gupta, D. J. Adams and N. G. Berry, Chem. Sci., 2016, 7, 4713–4719 RSC.
- F. Li, J. Han, T. Cao, W. Lam, B. Fan, W. Tang, S. Chen, K. L. Fok and L. Li, Proc. Natl. Acad. Sci. U. S. A., 2019, 116, 11259 CrossRef CAS PubMed.
- P. W. J. M. Frederix, G. G. Scott, Y. M. Abul-Haija, D. Kalafatovic, C. G. Pappas, N. Javid, N. T. Hunt, R. V. Ulijn and T. Tuttle, Nat. Chem., 2014, 7, 30 CrossRef PubMed.
- J. H. van Esch, Langmuir, 2009, 25, 8392–8394 CrossRef CAS PubMed.
- X. Du, J. Zhou, J. Shi and B. Xu, Chem. Rev., 2015, 115, 13165–13307 CrossRef CAS PubMed.
- K. McAulay, B. Dietrich, H. Su, M. T. Scott, S. Rogers, Y. K. Al-Hilaly, H. Cui, L. C. Serpell, A. M. Seddon, E. R. Draper and D. J. Adams, Chem. Sci., 2019, 10, 7801–7806 RSC.
- Y. Wang, W. Qi, J. Wang, Q. Li, X. Yang, J. Zhang, X. Liu, R. Huang, M. Wang, R. Su and Z. He, Chem. Mater., 2018, 30, 7902–7911 CrossRef CAS.
- E. R. Draper, B. Dietrich, K. McAulay, C. Brasnett, H. Abdizadeh, I. Patmanidis, S. J. Marrink, H. Su, H. Cui, R. Schweins, A. Seddon and D. J. Adams, Matter, 2020 DOI:10.1016/j.matt.2019.12.028.
- J. N. Israelachvili, D. J. Mitchell and B. W. Ninham, J. Chem. Soc., Faraday Trans. 2, 1976, 72, 1525–1568 RSC.
- C. Liu, Y. Wang, Y. Gao, Y. Zhang, L. Zhao, B. Xu and L. S. Romsted, Phys. Chem. Chem. Phys., 2019, 21, 8633–8644 RSC.
- N. Vlachy, M. Drechsler, J.-M. Verbavatz, D. Touraud and W. Kunz, J. Colloid Interface Sci., 2008, 319, 542–548 CrossRef CAS PubMed.
- L. Chen, S. Revel, K. Morris, L. C. Serpell and D. J. Adams, Langmuir, 2010, 26, 13466–13471 CrossRef CAS PubMed.
- C. Tang, A. M. Smith, R. F. Collins, R. V. Ulijn and A. Saiani, Langmuir, 2009, 25, 9447–9453 CrossRef CAS PubMed.
- A. Z. Cardoso, L. L. E. Mears, B. N. Cattoz, P. C. Griffiths, R. Schweins and D. J. Adams, Soft Matter, 2016, 12, 3612–3621 RSC.
- E. Rodd Lucy, P. Scott Timothy, J. Cooper-White Justin and H. McKinley Gareth, Appl. Rheol., 2005, 15, 12 Search PubMed.
- E. R. Draper, H. Su, C. Brasnett, R. J. Poole, S. Rogers, H. Cui, A. Seddon and D. J. Adams, Angew. Chem., Int. Ed., 2017, 56, 10467–10470 CrossRef CAS PubMed.
- A. Rüter, S. Kuczera, D. J. Pochan and U. Olsson, Langmuir, 2019, 35, 5802–5808 CrossRef PubMed.
- M. Wallace, J. A. Iggo and D. J. Adams, Soft Matter, 2017, 13, 1716–1727 RSC.
- G. R. Fulmer, A. J. M. Miller, N. H. Sherden, H. E. Gottlieb, A. Nudelman, B. M. Stoltz, J. E. Bercaw and K. I. Goldberg, Organometallics, 2010, 29, 2176–2179 CrossRef CAS.
- D. J. Adams, M. F. Butler, W. J. Frith, M. Kirkland, L. Mullen and P. Sanderson, Soft Matter, 2009, 5, 1856–1862 RSC.
- Y. Pocker and E. Green, J. Am. Chem. Soc., 1973, 95, 113–119 CrossRef CAS PubMed.
- A. J. Engler, S. Sen, H. L. Sweeney and D. E. Discher, Cell, 2006, 126, 677–689 CrossRef CAS PubMed.
- S. Roy, N. Javid, J. Sefcik, P. J. Halling and R. V. Ulijn, Langmuir, 2012, 28, 16664–16670 CrossRef CAS PubMed.
- S. Roy, N. Javid, P. W. J. M. Frederix, D. A. Lamprou, A. J. Urquhart, N. T. Hunt, P. J. Halling and R. V. Ulijn, Chem. – Eur. J., 2012, 18, 11723–11731 CrossRef CAS PubMed.
- L. Chen, G. Pont, K. Morris, G. Lotze, A. Squires, L. C. Serpell and D. J. Adams, Chem. Commun., 2011, 47, 12071–12073 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0cc01252a |
| This journal is © The Royal Society of Chemistry 2020 |