
Air and water stable secondary phosphine oxides as diazaphospholene precatalysts†
Travis
Lundrigan
a,
Chieh-Hung
Tien
a,
Katherine N.
Robertson
 b and
Alexander W. H.
Speed
*a
b and
Alexander W. H.
Speed
*a
aDepartment of Chemistry, Dalhousie University, Halifax, Nova Scotia B3H 4R2, Canada. E-mail: aspeed@dal.ca
bDepartment of Chemistry, Saint Mary's University, Halifax, Nova Scotia B3H 3C3, Canada
First published on 4th March 2020
Abstract
Air-stable secondary phosphine oxides (SPOs) are readily formed from diazaphospholene bromides. In the presence of pinacolborane, these SPOs are transformed into catalytically active diazaphospholene hydrides. A silyl triflate transforms the SPOs into phosphenium triflates. The use of diazaphospholene SPOs as reduction reaction precatalysts was validated by imine reduction, conjugate reduction, pyridine hydroboration, and asymmetric reduction.
Diazaphospholenes have recently emerged as a class of catalyst for reduction reactions. Gudat showed that highly air-sensitive diazaphospholene hydrides were stoichiometric reducing reagents for aldehydes, ketones, and conjugate acceptors.1 Kinjo showed that ammonia borane could regenerate a diazaphospholene hydride in the reduction of an azo compound, allowing catalysis with diazaphospholenes.2a Kinjo then showed that pinacolborane (HB(pin)) and diphenylsilane could be terminal reductants in aldehyde and carbon dioxide reductions, respectively. Importantly, pinacolborane transformed an alkoxydiazaphospholene to a diazaphospholene hydride in Kinjo's work (eqn (1), Fig. 1).2b,c Our research group has exploited the alkoxydiazaphospholene intermediate from Kinjo's aldehyde-reduction catalytic cycle as a precatalyst for diazaphospholenes. We used this system to catalyze imine reductions and render Gudat's conjugate reductions catalytic.3a Kinjo and our group separately reported that diazaphospholenes catalyze the reduction of pyridines, with the distinct difference that our group used a neutral precatalyst, while Kinjo used a cationic triflate.4 We subsequently employed the pre-catalyst strategy to form a chiral diazaphospholene for imine reduction.3b Cramer disclosed an elegant chiral diazaphospholene precatalyst based on an alkoxydiazaphospholene for conjugate reduction and Claisen reactions.5 Other reported systems relevant to diazaphospholene pre-catalysts include Radosevich's trisamidophosphine that forms a diazaphospholene in the presence of HB(pin),6 and diazaarsolenes bearing alkoxy substituents reported by Melen. Arsenic hydrides have not been isolated from these systems, however these arsenic catalysts do engage in reduction catalysis in analogy to the dizazphospholene systems.7 Yang and Cheng showed that diazaphospholenes are the most hydridic neutral compounds yet quantified on the Mayr nucleophilicity scale, promising a rich future for diazaphospholenes as reduction catalysts and reagents.8
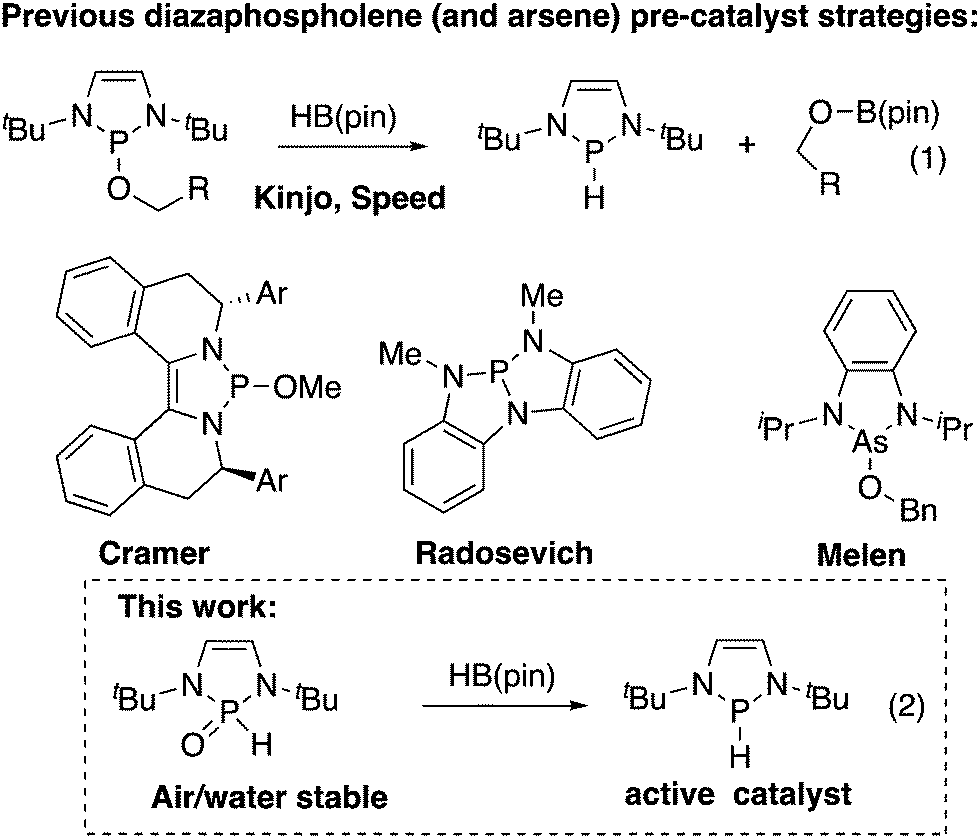 | ||
| Fig. 1 Comparison of diazaphospholene precatalysts. | ||
With this intense interest in diazaphospholenes, further developments to increase the accessibility of these catalysts in organic synthesis is warranted. This work shows that the secondary phosphine oxide (SPO) hydrolysis products of diazaphospholene alkoxides or halides are air and water stable, can be purified by silica gel chromatography, and can serve as precatalysts for diazaphospholene hydrides (eqn (2), Fig. 1) and the corresponding phosphenium triflates.
During our initial investigations of diazaphospholenes, we observed that samples of diazaphospholene precatalyst 1a in standard NMR tubes hydrolyzed to SPO 2a because of the incursion of water (Scheme 1).3 SPOs of diazaphospholenes have been rationally synthesized as ligands for metals via hydrolysis of amidodiazaphospholene 1b.9 We also noted that when HB(pin) was present with 1a, SPO formation was not observed on the same timeframe. We considered that HB(pin) could either be scavenging the water or could be reducing the SPO. While this work was in progress, Webster and co-workers reported the reduction of dialkyl and diaryl SPOs directly to uncomplexed phosphines using pinacolborane at room temperature.10
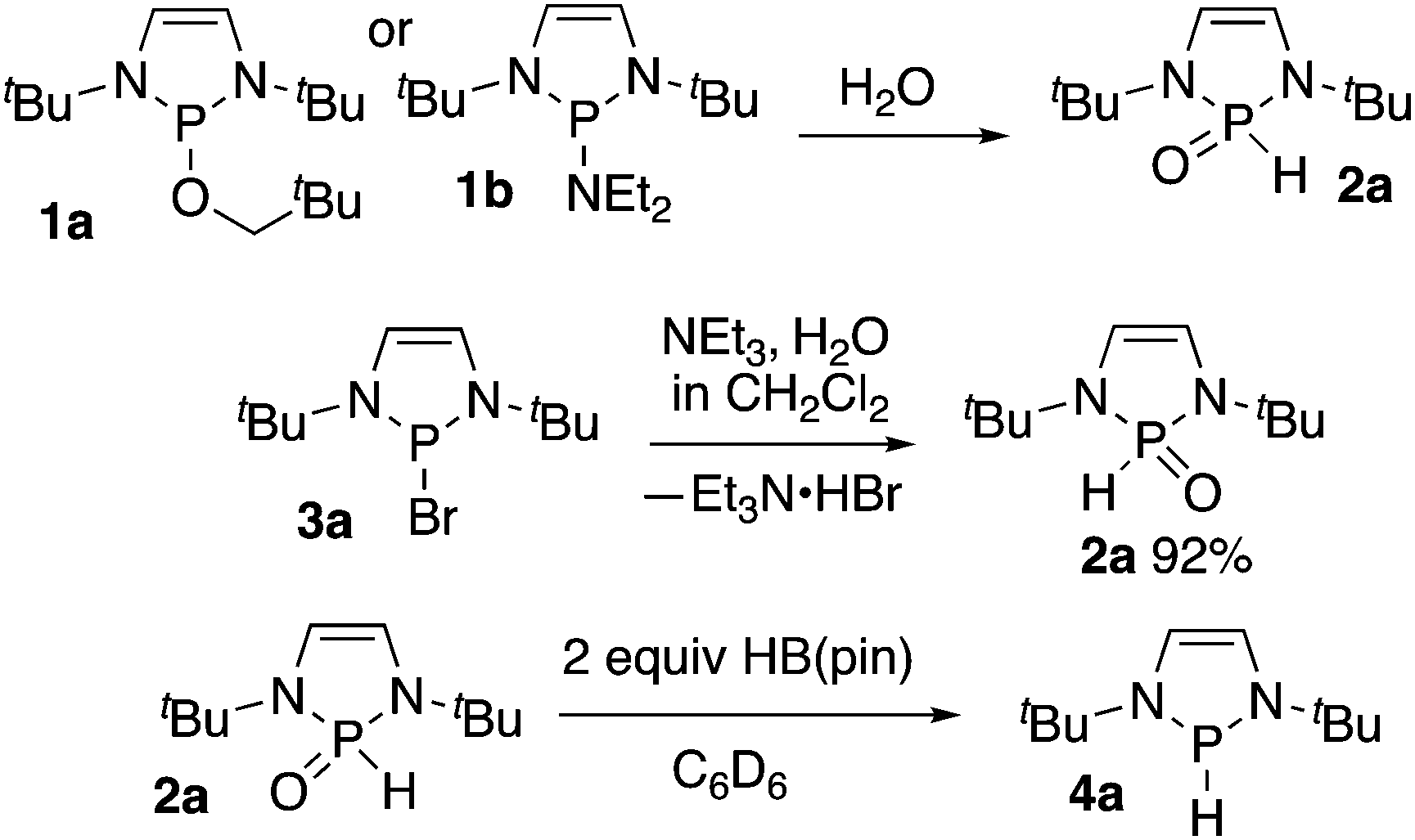 | ||
| Scheme 1 Synthesis of a diazaphospholene SPO, and its transformation to hydride. | ||
In a more direct synthesis of 2a we avoided the preparation of intermediate 1a or 1b by combining diazaphospholene bromide 3a with triethylamine in dichloromethane followed by addition of water to afford SPO 2a. Compound 2a is a solid that was stable to column chromatography in air (Scheme 1). NMR spectral data matched that previously reported for 2a.9a Additionally, a crystal of 1a grown from ethyl acetate/hexanes has the same unit cell as that previously reported for crystals of 2a.9a,b HB(pin) transforms 2a to hydride 4a in various solvents, including benzene-d6, and THF. While 1 equivalent of HB(pin) was sufficient for the transformation, the reaction proceeded significantly faster when greater than 1 equivalent of HB(pin) was used.
We used 2a in reductions employing HB(pin) that are known to be catalyzed by diazaphospholenes, and have been documented to not undergo an uncatalyzed reaction with HB(pin) to validate the function of 2a as a precatalyst (Scheme 2). Imine 5a was reduced to the corresponding amine with HB(pin) in the presence of 1 mol% of 2a.3a Chalcone 6 was reduced to compound 7 with 1 mol% of 2a.3a,5a,11 Precatalyst 2a also effected the reduction of pyridines with HB(pin). Nicotinonitrile 8a was reduced to dihydropyridine 9a, while 3-acetylpyridine 8b was reduced to dihydropyridine 9b without reduction of the ketone. These results show similar chemoselectivity and selectivity to previous pyridine reductions performed with precatalyst 1a.4
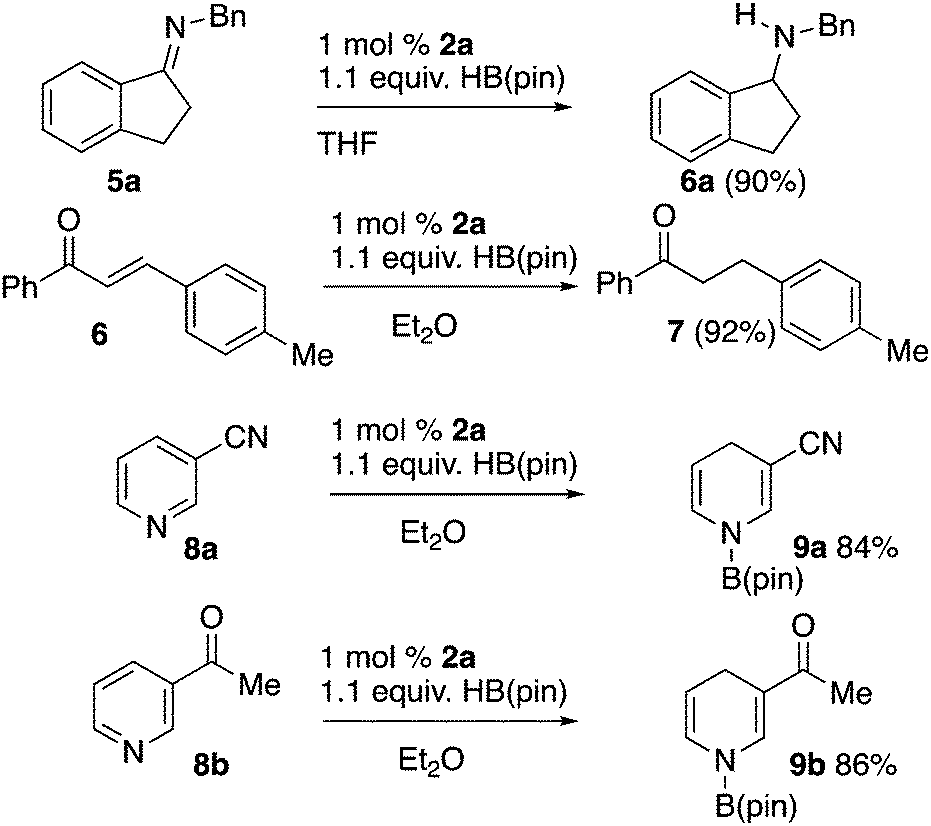 | ||
| Scheme 2 Reduction reactions with SPO precatalyst 2a. | ||
We subsequently explored the chemistry of a chiral SPO precatalyst (Scheme 3). Compound 2b was prepared from the hydrolysis of the corresponding bromide and was also an air and water stable substance that could be purified by silica gel chromatography.3b In a further simplification of the procedure, isolation of 3b could be completely avoided. Diimine 10 was cyclized with PBr3 and cyclohexene according to MacDonald and co-workers’ procedure for diazaphospholene generation.12 After one hour, triethylamine and water were added directly to the reaction mixture. After aqueous work-up and chromatography, 2b was obtained in good yield. This procedure allowed preparation of 2b in one pot from starting material 10, without the isolation of any air-sensitive intermediates. Prior NMR spectroscopy of 3b had shown equivalence of the naphthylethyl sidechains and backbone protons, implying a C2 symmetric structure, which strongly suggests that the P–Br bond is ionized in solution, resulting in planarization of the phosphorus.3b,13 NMR spectroscopy of 2b shows less symmetry: the naphthylethyl side-chains and backbone protons of the diazaphospholene are now inequivalent, a consequence of the breaking of C2 symmetry because of the distinct H and O phosphorus substituents in the SPO. X-ray crystallography confirmed the structure of 2b.14
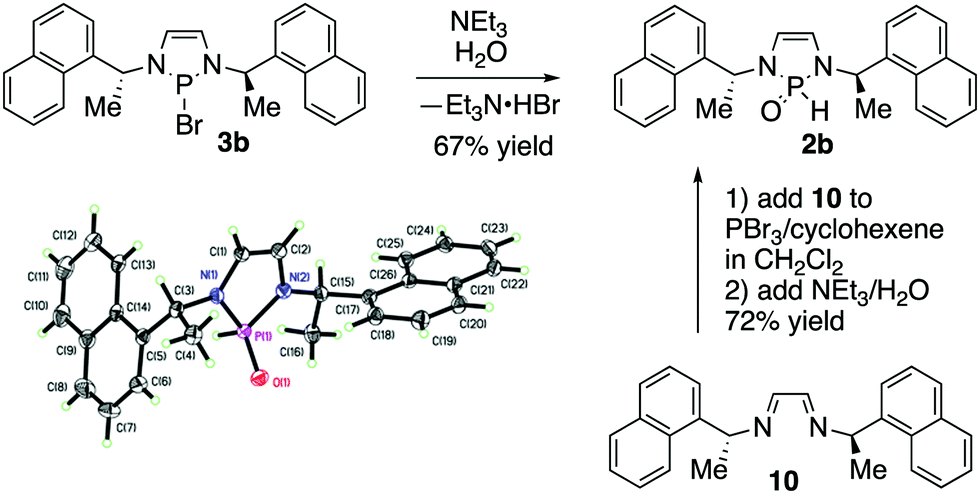 | ||
| Scheme 3 Preparation and X-ray structure of chiral SPO precatalyst 2b. | ||
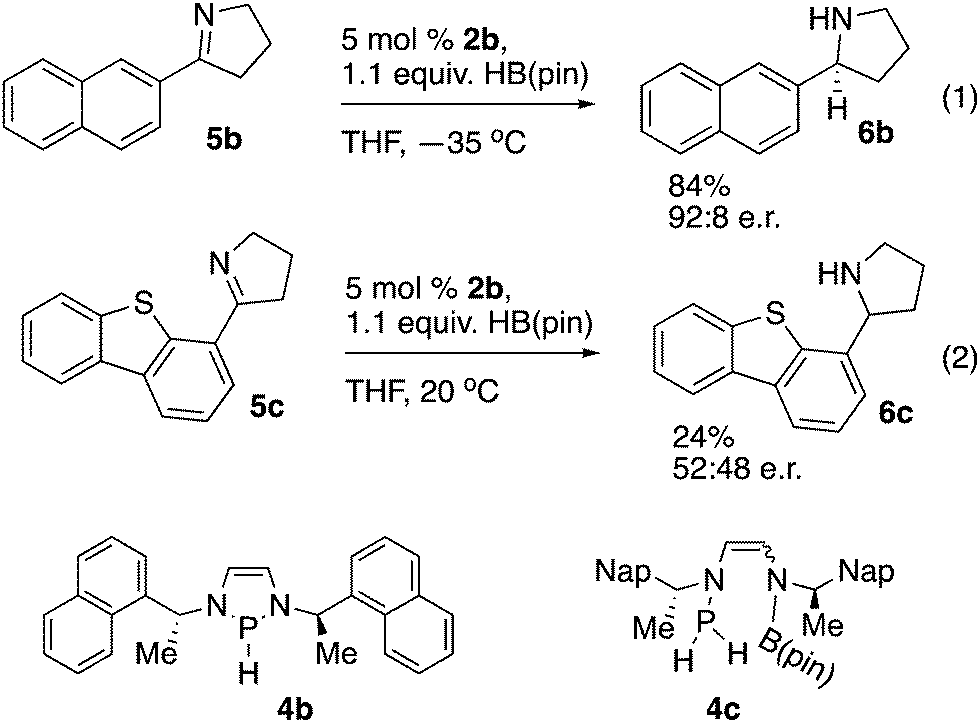
Asymmetric reduction reactions were explored with chiral precatalyst 2b (Scheme 4). Reduction of imine 5b to amine 6b (eqn (1), Scheme 4) proceeded with comparable enantioselectivity to that previously observed with chiral diazaphospholenes with an 85![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :15 e.r. at room temperature, and an increase to 92:8 e.r. at −35 °C.16 A limitation for the use of 2b was revealed when sterically hindered imine 5c gave a low yield of essentially racemic product 6c under these conditions (eqn (2), Scheme 4). To further understand these distinctive results, we repeated the experiments in NMR tubes and observed the reaction progress by 11B and 31P NMR spectroscopy. After the components were mixed, the reaction of eqn (1) showed almost complete consumption of HB(pin) according to the 11B NMR spectrum after 5 minutes. The 31P NMR spectrum of this reaction showed a mixture of pre-catalyst 2b and hydride 4b. The reaction of eqn (2) exhibited substantially different behavior. The 11B NMR spectrum showed essentially no conversion after both 5 minutes and two hours, with mostly unreacted HB(pin) present. At both the 5 minute and two hour mark, the 31P NMR spectrum of the reaction of eqn (2) showed a relatively complex mixture of phosphorus containing products, including putative 4c, and PH3. These endocyclic cleavage products have previously been noted in the decomposition of diazaphospholenes, and do not appear to be catalytically active in imine reduction.3 Accordingly, with a readily reduced substrate such as 5b, pre-catalyst activation and imine reduction occur, while with a challenging substrate such as 5c, reduction does not occur before decomposition of the catalyst. The observed formation of 6c in the reaction of eqn (2) is presumably due to an uncatalyzed reduction of the imine with HB(pin) mediated by the water introduced during the quenching of the reaction.15
:15 e.r. at room temperature, and an increase to 92:8 e.r. at −35 °C.16 A limitation for the use of 2b was revealed when sterically hindered imine 5c gave a low yield of essentially racemic product 6c under these conditions (eqn (2), Scheme 4). To further understand these distinctive results, we repeated the experiments in NMR tubes and observed the reaction progress by 11B and 31P NMR spectroscopy. After the components were mixed, the reaction of eqn (1) showed almost complete consumption of HB(pin) according to the 11B NMR spectrum after 5 minutes. The 31P NMR spectrum of this reaction showed a mixture of pre-catalyst 2b and hydride 4b. The reaction of eqn (2) exhibited substantially different behavior. The 11B NMR spectrum showed essentially no conversion after both 5 minutes and two hours, with mostly unreacted HB(pin) present. At both the 5 minute and two hour mark, the 31P NMR spectrum of the reaction of eqn (2) showed a relatively complex mixture of phosphorus containing products, including putative 4c, and PH3. These endocyclic cleavage products have previously been noted in the decomposition of diazaphospholenes, and do not appear to be catalytically active in imine reduction.3 Accordingly, with a readily reduced substrate such as 5b, pre-catalyst activation and imine reduction occur, while with a challenging substrate such as 5c, reduction does not occur before decomposition of the catalyst. The observed formation of 6c in the reaction of eqn (2) is presumably due to an uncatalyzed reduction of the imine with HB(pin) mediated by the water introduced during the quenching of the reaction.15
 | ||
| Scheme 4 Asymmetric catalysis with SPO precatalyst 2b. | ||
Our recently reported cationic diazaphospholene catalysts are able to reduce substrate 5c in a highly enantioselective manner, showing that 2b has inferior reactivity.16
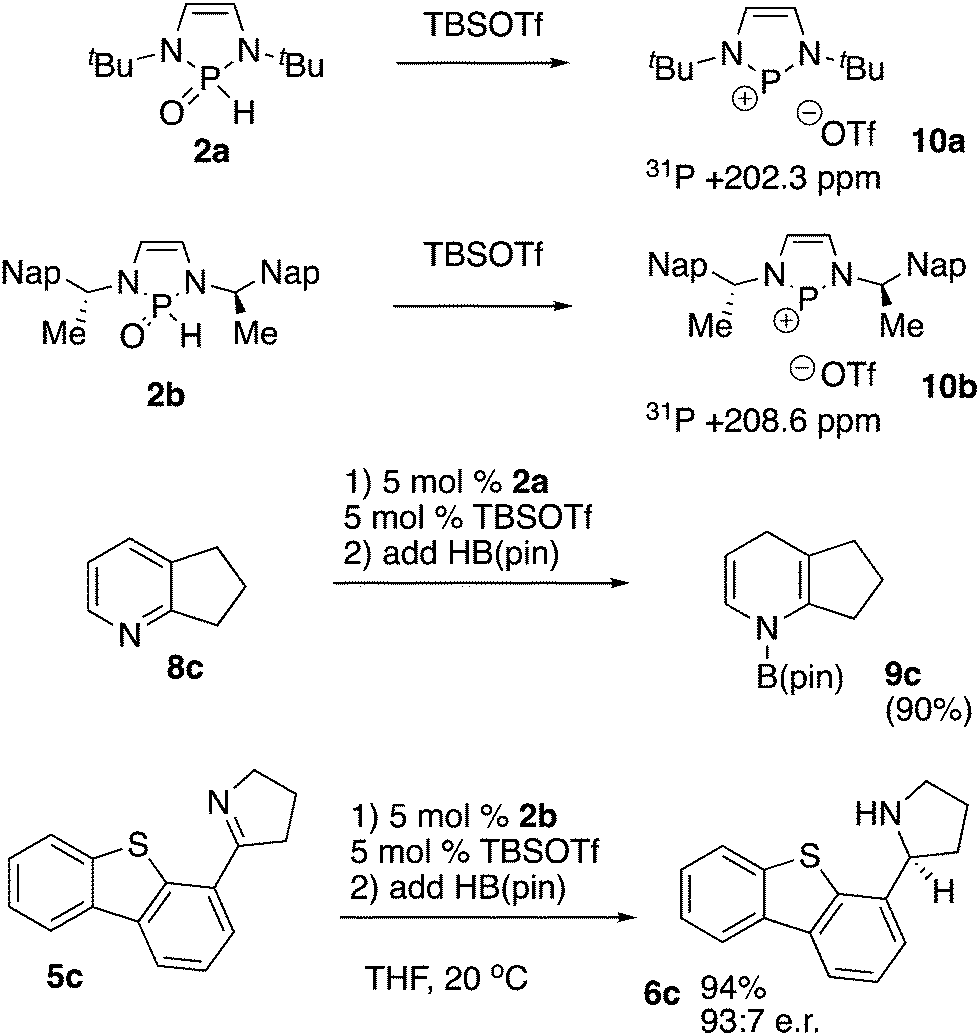
Given this limitation, we sought to determine if 2a and 2b could be used to generate phosphenium cations in situ, enabling access to more active phosphenium triflate reduction catalysts. Exposure of 2a or 2b to TBS triflate in dry chloroform-d on an NMR scale immediately resulted in the formation of phosphenium ions 10a and 10b, as evidenced by appearance of a signal in the 31P NMR spectrum at +202.3 ppm for 2a, and 208.6 ppm for 2b (Scheme 5). Additionally, the formerly diastereotopic naphthylethyl groups in 2b became equivalent in the 1H NMR spectrum, indicating the generation of C2 symmetry by planarization of the phosphorus centre. The mixture containing 2a proved competent in the reduction of pyridine 8c to the dihydropyridine 9c. Pyridines such as 8c with no electron withdrawing group would not be expected to readily undergo reduction with neutral diazaphospholenes.5b Kinjo has effected reductions of such pyridines employing phosphenium cations.5a In the chiral system, exposure of 2b to TBS triflate, followed by addition of imine 5c, and HB(pin) restored asymmetric induction with 5c, leading to selective formation of 6c. This contrasts with the absence of selectivity observed with precatalyst 2b for the reduction of 5c in the absence of the silyl triflate. The observed reactivity and selectivity are close to that previously observed in the reduction of compound 5c catalyzed by isolated phosphenium ion 10b, and it would be expected that the large scope of substrates reduced by isolated 10b could also be prepared under these in situ conditions. In addition, endocyclic cleavage of 10b with excess HB(pin) has never been noted, enabling the reduction of more challenging substrates such as 5c to compete with catalyst decomposition.16
 | ||
| Scheme 5 Catalysis with phosphenium cations generated from SPO precatalysts 2a and 2b. | ||
In conclusion, we have validated the use of SPOs derived from diazaphospholenes as precatalysts for the catalytically active diazaphospholene hydrides. Given growing interest in diazaphospholenes, the demonstration that secondary phosphine oxides can serve as bench-purifiable and air stable precatalysts will further help to popularize diazaphospholene chemistry. The stability of these entities to chromatography will enable the separation of diazaphospholene precursors from complex product mixtures, potentially allowing access to diazaphospholenes that cannot currently be accessed cleanly. We have also demonstrated generation of phosphenium cations from diazaphospholene SPOs, showing how these highly active asymmetric reduction catalysts can also be accessed from air-stable precursors.
Financial support from NSERC (Discovery Grant and I2I Grant), the Killam Foundation (C.-H. T.) are acknowledged. Dr Mike Lumsden and Mr Xiao Feng are thanked for assistance with NMR spectroscopy and mass spectrometry, respectively.
Conflicts of interest
The authors declare the following competing financial interest(s): Dalhousie University has filed patents on chiral phosphenium triflates and secondary phosphine oxides for asymmetric imine reduction, from which royalty payments may be derived.References
- (a) D. Gudat, A. Haghverdi and M. Nieger, Angew. Chem., Int. Ed., 2000, 39, 3084 CrossRef CAS; (b) S. Burck, D. Gudat, M. Nieger and W. W. Du Mont, J. Am. Chem. Soc., 2006, 128, 3946 CrossRef CAS PubMed; (c) D. Gudat, Dalton Trans., 2016, 45, 5896 RSC; (d) D. Gudat, Diazaphospholene Chemistry. In Encyclopedia of Inorganic and Bioinorganic Chemistry, Online, ed. R. A. Scott, John Wiley and Sons, Hoboken, NJ, 2nd edn, 2018 Search PubMed.
- (a) C. C. Chong, H. Hirao and R. Kinjo, Angew. Chem., Int. Ed., 2014, 53, 3342 CrossRef CAS PubMed; (b) C. C. Chong, H. Hirao and R. Kinjo, Angew. Chem., Int. Ed., 2015, 54, 190 CrossRef CAS PubMed; (c) C. C. Chong and R. Kinjo, Angew. Chem., Int. Ed., 2015, 53, 12116 CrossRef PubMed.
- (a) M. R. Adams, C.-H. Tien, B. S. N. Huchenski, M. J. Ferguson and A. W. H. Speed, Angew. Chem., Int. Ed., 2017, 56, 6268 CrossRef CAS PubMed; (b) M. R. Adams, C.-H. Tien, R. McDonald and A. W. H. Speed, Angew. Chem., Int. Ed., 2017, 56, 16660 CrossRef CAS PubMed.
- (a) B. Rao, C. C. Chong and C. R. Kinjo, J. Am. Chem. Soc., 2018, 140, 652 CrossRef CAS PubMed; (b) T. Hynes, E. N. Welsh, R. McDonald, M. J. Ferguson and A. W. H. Speed, Organometallics, 2018, 37, 841 CrossRef CAS.
- (a) S. Miaskiewics, J. H. Reed, P. A. Donets, C. C. Oliveira and N. Cramer, Angew. Chem., Int. Ed., 2018, 57, 4039 CrossRef PubMed; (b) J. H. Reed, P. A. Donets, S. Miaskiewicz and N. A. Cramer, Angew. Chem., Int. Ed., 2019, 58, 8893 CrossRef CAS PubMed.
- Y.-C. Lin, E. Hatzakis, S. M. McCarthy, K. D. Reichl, T.-Y. Lai, H. P. Yennawar and A. T. Radosevich, J. Am. Chem. Soc., 2017, 139, 6008 CrossRef CAS PubMed.
- D. M. C. Ould and R. L. Melen, Chem. – Eur., J., 2018, 24, 15201 CrossRef CAS PubMed.
- J. Zhang, J.-D. Yang and J.-P. Cheng, Angew. Chem., Int. Ed., 2019, 58, 5983 CrossRef CAS PubMed.
- (a) Y.-C. Chang, Y.-C. Lee, M.-F. Chang and F.-E. Hong, J. Organomet. Chem., 2016, 808, 23–33 CrossRef CAS; (b) CSD refcode TABTAU.
- C. B. Provis-Evans, E. A. C. Emanuelsson and R. L. Webster, Adv. Synth. Catal., 2018, 360, 3999 CrossRef CAS.
- C. C. Chong, B. Rao and R. Kinjo, ACS Catal., 2017, 7, 5814 CrossRef CAS.
- J. W. Dube, G. J. Farrar, E. L. Norton, K. L. S. Szekeley, B. F. T. Cooper and C. L. B. Macdonald, Organometallics, 2009, 28, 4377 CrossRef CAS.
- S. Burck, D. Gudat, M. Nieger, Z. Benkö, L. Nyulászi and D. Szieberth, Z. Anorg. Allg. Chem., 2009, 635, 245 CrossRef CAS.
- Compound 2b was crystalized from ethyl acetate/hexanes as a 1:1 solvate with ethyl acetate. The ethyl acetate is not shown in the ORTEP in Scheme 3 for clarity.
- B. S. N. Huchenski and A. W. H. Speed, Org. Biomol. Chem., 2019, 17, 1999 RSC.
- T. L. Lundrigan, E. N. Welsh, T. Hynes, C.-H. Tien, M. R. Adams, K. R. Roy, K. N. Robertson and A. W. H. Speed, J. Am. Chem. Soc., 2019, 141, 14083 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1981976. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0cc01072c |
| This journal is © The Royal Society of Chemistry 2020 |