
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Conjugated polymer donor–molecular acceptor nanohybrids for photocatalytic hydrogen evolution†
Haofan
Yang
,
Xiaobo
Li
 *,
Reiner Sebastian
Sprick
and
Andrew I.
Cooper
*
*,
Reiner Sebastian
Sprick
and
Andrew I.
Cooper
*
Department of Chemistry and Materials Innovation Factory, University of Liverpool, 51 Oxford Street, Liverpool L7 3NY, UK. E-mail: Xiaobo.li@liverpool.ac.uk; aicooper@liverpool.ac.uk
First published on 19th May 2020
Abstract
A library of 237 organic binary/ternary nanohybrids consisting of conjugated polymers donors and either fullerene or non-fullerene molecular acceptors was prepared and screened for sacrificial photocatalytic hydrogen evolution activity. PCDTBT/PC60BM nanohybrid showed a high hydrogen evolution rate of 105.2 mmol g−1 h−1 under visible light (λ > 420 nm).
Direct photocatalytic solar hydrogen production promises a technologically simple way to convert solar energy into chemical fuels.1 To be scalable, however, this approach requires more efficient photocatalysts. Inorganic semiconductor photocatalysts have been explored widely for some time.2,3 Recently, organic photocatalysts have also attracted attention due to potential advantages in terms of tunable composition, structure, and properties.4 These organic photocatalysts include polymeric carbon nitride,5–7 carbon dots,8,9 conjugated microporous polymers,10–12 covalent triazine-based frameworks13,14 and covalent organic frameworks.15–18 However, organic photocatalysts also have some inherent drawbacks, such as strong exciton binding energies, low charge-carrier mobilities, and short charge migration pathlengths. In organic photovoltaics (OPV), these issues have been mitigated by the introduction of donor/acceptor bulk heterojunctions.19 Such nanoscale blends of a donor and an acceptor ensures that excitons can reach an interface and dissociate into free charge carriers. In principle, this concept should also be transferable to organic polymer photocatalysts.
Here, we designed a library of donor–acceptor nanohybrids (DANHs) photocatalysts that combine conjugated polymer donors with either fullerene/non-fullerene molecular acceptors (Fig. 1). We then evaluated their photocatalytic hydrogen evolution performance. Organic nanoparticles composed of a single conjugated polymer were developed previously for photocatalytic hydrogen production,20–22 but those photocatalysts showed rapid deactivation (in less than 2 hours) after a high initial hydrogen evolution rate of 52.4 mmol g−1 h−1 (17 μmol h−1). By comparison, the organic DANHs photocatalysts reported here show both increased hydrogen evolution rates (up to 179.0 mmol g−1 h−1) and enhanced photocatalytic stability (sustained H2 production for at least 18 hours).
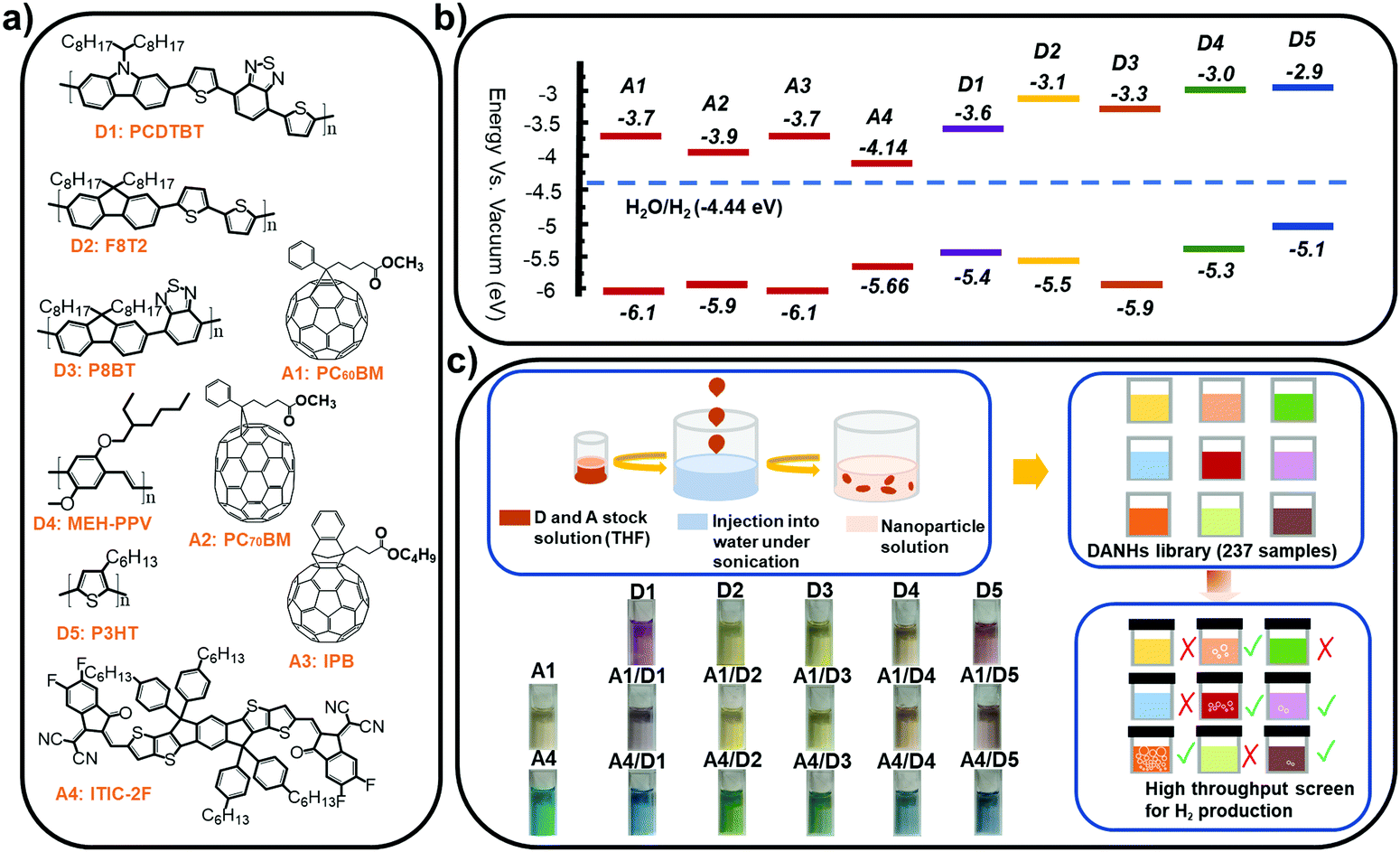 | ||
| Fig. 1 (a) Chemical structures and (b) energy levels of the conjugated polymer donors and molecular acceptors studied here (obtained from commercial supplier Ossila except D5 obtained from ref. 25). (c) Nano-precipitation process used to prepare the DANHs and scheme representing high-throughput screening process for photocatalytic activity. Photographs show DANHs aqueous solutions in cuvettes. | ||
We studied five conjugated polymer donors (D1–D5) and four molecular acceptors (A1–A4) (Fig. 1a and b). Water dispersible DANHs were prepared using nano-precipitation strategy,23 as shown in Fig. 1c. A tetrahydrofuran (THF) solution containing the polymer donor and the molecular acceptor was injected into water with continuous sonication, followed by the evaporation of the THF (detailed procedures in ESI†). Colloidal solutions of DANHs were obtained (Fig. 1c) after THF removal. It is well known that the composition ratio between donor–acceptor is critical for photovoltaic performance. Likewise here for photocatalysis, we screened a broad range of relative donor-to-acceptor ratios to give a total DANH library of 237 samples. The photocatalytic hydrogen evolution performance of this library was then screened using high-throughput parallel 48-sample photocatalysis screen, as introduced previously by our group.24Fig. 2 plots the sacrificial photocatalytic hydrogen evolution rate for A1–A4/D1–D5 DANH combinations as a function of donor/acceptor composition ratios (w/w%). The photocatalytic performance of the DANHs is strongly dependent on this ratio.
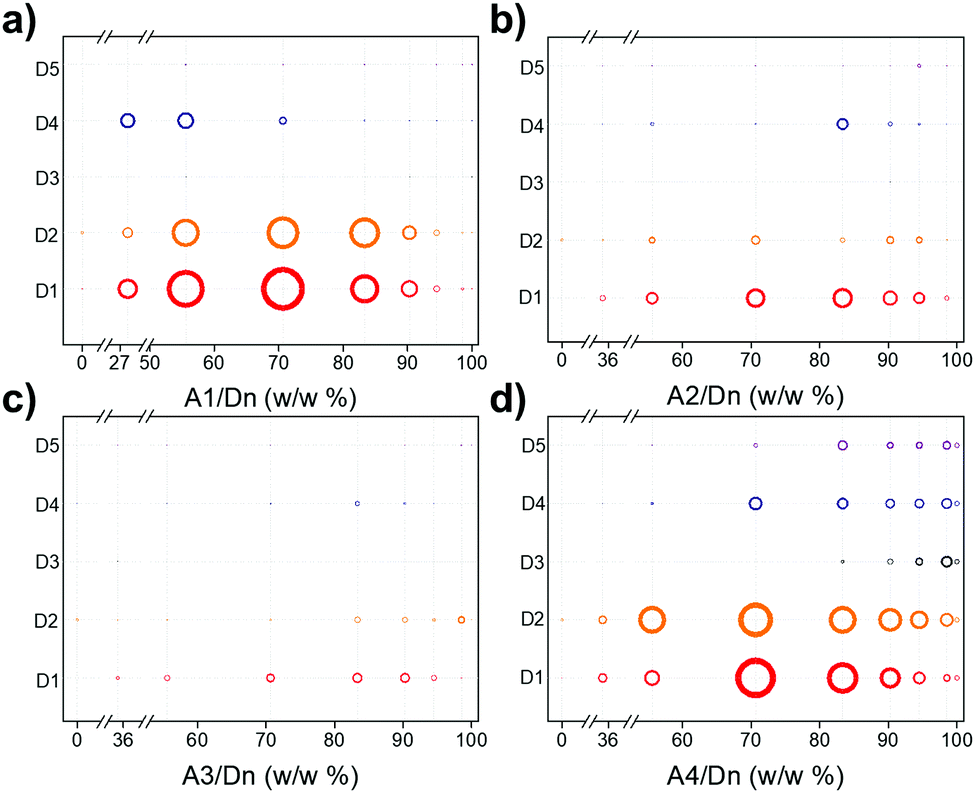 | ||
Fig. 2 Sacrificial photocatalytic hydrogen production activities for the various combinations of polymer donors and molecular acceptors: (a) A1/Dn; (b) A2/Dn; (c) A3/Dn, and; (d) A4/Dn, plotted as a function of the acceptor weight fraction that was added in the nanoprecipitation process (100% corresponds to the pure acceptor nanoparticle). Testing conditions: catalyst concentration = 20–100 μg mL−1 (0.1–0.5 mg in 5 mL water); ascorbic acid (0.04 M); Pt loading based on total mass of donor and acceptor: (3 wt% using a stock solution of H2PtCl6, 8 wt% in water); light source = solar simulator, 1 sun; irradiation time = 2 hours. The hydrogen evolution rate is proportional to the area of the circles (for scale, the maximum hydrogen evolution rate found was 171.4 mmol g−1 h−1 for 70.6![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :29.4 w/w% DANH of A1/D4 (Fig. 2d)). :29.4 w/w% DANH of A1/D4 (Fig. 2d)). | ||
DANHs combinations that involved A3 showed little improvement in hydrogen evolution rate (HER) with respect to the pure donor polymers or molecular acceptors (Fig. 2c). By contrast, combinations involving A2 showed synergies at acceptor ratios in the range 70–80 w/w, particular for polymer D1 (Fig. 2b). However, much higher HERs were observed for DANHs that included A1 (Fig. 2a) and A4 (Fig. 2d). The most promising combinations were found to be A1/D1 and A1/D2 (Fig. 2a) and A4/D1, and A4/D2 (Fig. 2d).
For the A1 series, the highest performance, 165.2 ± 25.7 mmol g−1 h−1, was observed for A1/D1 DANHs at a mass ratio of 70.6/29.4. This HER is 55 times higher than the HER for the pure acceptor, A1 (3.0 ± 0.7 mmol g−1 h−1), and 165 times higher than the pure polymer donor, D1 (1.0 ± 0.4 mmol g−1 h−1), respectively, showing a strong synergistic effect in these DANHs. The results of a more exhaustive search of the variation of HER with A1/D1 composition are shown in Fig. S1 (ESI†), which substantiates the conclusions of the high-throughput screening (i.e., around 70 w/w% A1 gives the maximum HER). Combining polymer D1 with PC70BM (A2) (Fig. 2b) or [60]IPB (A3) (Fig. 2c) gives the same compositional trend in HER (a maximum at around 70–80 w/w% acceptor), but the HERs are much lower than observed for PC60BM (A1) (Fig. 2a).
Recently, high-performing non-fullerene based molecular acceptors have surpassed the most efficient fullerene acceptors for organic photovoltaics,26,27 which inspired us to prepare DANH photocatalysts using ITIC-2F (A4) as non-fullerene based acceptor. As shown in Fig. 2d, A4/D1 (70.6 wt% A4) exhibited the highest HER of 166.8 ± 7.9 mmol g−1 h−1 among the combinations in this library.
The coprecipitation of both donor and acceptor is important for the HER: physical mixtures of A1 and D1 nanoparticles (detailed procedures in ESI†) showed significantly lower HERs compared with the A1/D1 and A4/D1 DANHs (Fig. S2 (ESI†); activities 23 and 18 times lower, respectively, when A1 mass ratio is 70.6%), suggesting that the formation of donor–acceptor junctions is essential. This was supported by photoluminescence results that show complete quenching for both A1/D1 and A4/D1 DANHs, which results from efficient charge transfer. By contrast, physical mixtures of the donors and acceptors showed incomplete quenching as a result of the poor junction formation (Fig. S3, ESI†).
A3 has the same energy level as A1, but much lower catalytic activity was observed for A3/D1 compared to A1/D1. It is worth noting that A3 itself shows no HER compared to A1 with a HER of 3.0 mmol g−1 h−1. There are several important factors that might affect the photocatalytic hydrogen production process when using different molecular acceptors, such as the phase distribution of the donor–acceptor junction, which is crucial for exciton dissociation and migration, and the dispersed state of the platinum cocatalysts formed on donor–acceptor nanocomposites. We plan to investigate the details of this in the future for selected systems.
We also studied the effect of ascorbic acid concentration, cocatalyst type, and cocatalyst loading on the HER for the A1/D1 NADH (70.6 wt% A1). The HER could be further improved to 179.0 mmol g−1 h−1 from 165.2 mmol g−1 h−1 (screening conditions) by using 0.2 M ascorbic acid and 3% Pt loading (Fig. S4, ESI†). Platinum was found to be the most efficient co-catalyst of a range of 11 catalyst precursors (Fig. S5, ESI†). We also tried to tune the morphology and size of the A1/D1 NADHs by introducing surfactants during nanoprecipitation (Fig. S6, ESI†), but this was found to markedly decrease the HER. No appreciable hydrogen generation could be detected for A1/D1 (70.6 wt% A1) in the absence the scavenger (ascorbic acid), Pt, or light irradiation.
Next, time-course photocatalytic hydrogen evolution rates for A1/D1 NADHs were investigated. A1/D1 NADHs showed an initial HER of 105.2 mmol g−1 h−1 (120.9 μmol h−1) in the first 2 hours under visible light illumination (λ > 420 nm) using condition 1 (Fig. 3a). Similar initial rates were observed for the other two sets of reaction conditions, which had different ascorbic acid concentrations and Pt loadings (Fig. 3a). The hydrogen evolution activity decreased over time, but the NADHs were still active after 18 hours photocatalysis, with a rate of 37.8 mmol g−1 h−1 over the last 4 hours. Therefore, the A1/D1 DANH both exhibits excellent H2 production rates and has much better stability compared with previously reported pure polymer nanoparticle photocatalysts,20–22 which show activity for only 1, 4 and 11 hours, respectively. The catalyst mass used here (1.15 mg) is also higher than for earlier studies (around 0.05 and 0.33 mg polymer).20,21
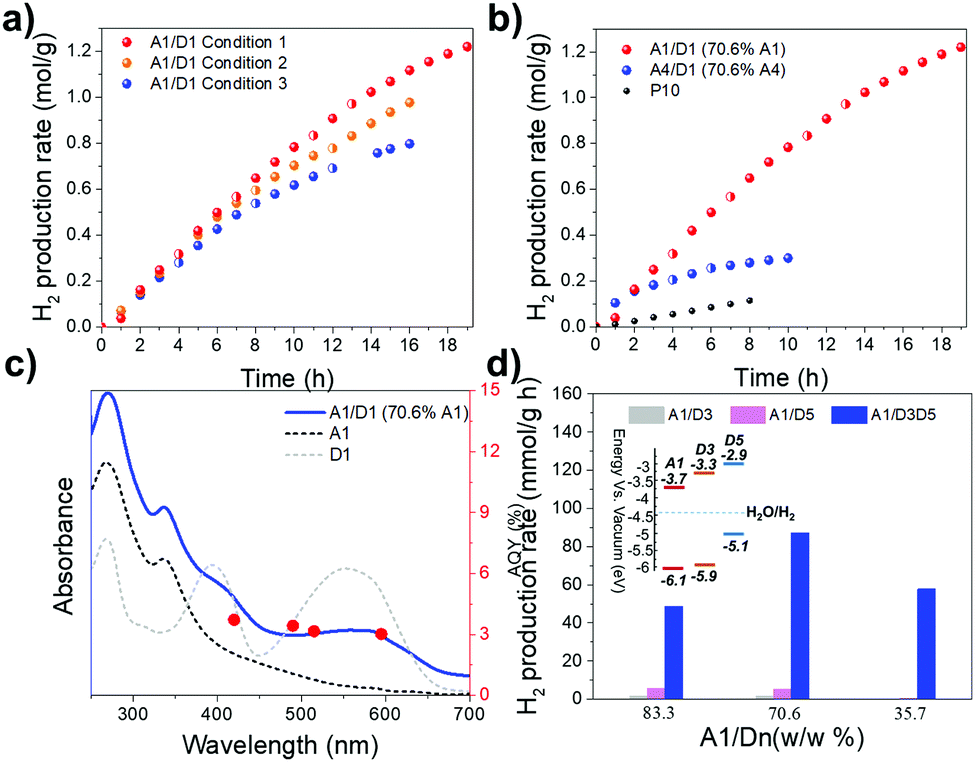 | ||
| Fig. 3 (a) Time course of hydrogen production for A1/D1 NADHs and a bulk, pure conjugated polymer, P10, irradiated by 300 W Xe lamp fitted with a λ > 420 nm filter using 1.15 mg of the catalyst. Condition 1: 0.1 M ascorbic acid and 9 wt% Pt; condition 2: 0.2 M ascorbic acid and 9 wt% Pt; condition 3: 0.2 M ascorbic acid and 3 wt% Pt. Half circle points represent the beginning of the next run after degassing. (b) Time course of hydrogen production for A1/D1 NADH, A4/D1 NADH, and bulk P10 irradiated by 300 W Xe lamp fitted with a λ > 420 nm filter using 1.15 mg of catalysts under condition 1. Half circle points represent the beginning of the next run after degassing. (c) UV-vis spectra and AQY of A1/D1 measured with monochromatic LED light at 420, 495, 515 and 595 nm, respectively. UV-vis spectrum of pure A1 and D1 nanoparticles are plotted with dashed lines (Intensity rescaled for clarity). (d) Hydrogen evolution rate of A1/D3:D5 ternary nanohybrids of various compositions. Catalyst concentrations: 0.3, 0.23 and 0.14 mg in 5 mL water; ascorbic acid: 0.04 M; Pt loading: 3 wt%; light source = solar simulator; irradiation time = 2 hours. Inset: HOMO and LUMO band levels of donors and acceptors. | ||
At lower catalysis loadings, even higher hydrogen evolution rates of 247.8 mmol g−1 h−1 and 383.4 mmol g−1 h−1 were observed for 0.23 mg and 0.115 mg of A1/D1 NADHs (70.6 wt% A1), respectively (Fig. S7, ESI†). Of course, for practical applications, the amount of hydrogen produced per unit area irradiated is the most important parameter, and hence such low catalyst loadings are less useful.
Compared to P10, one of the most efficient organic photocatalysts developed by our group,28 the A1/D1 DANH catalyst was almost 6 times more active in terms of mass-normalized rate over 8 hours (85.0 mmol g−1 h−1 for A1/D1 versus 14.3 mmol g−1 h−1 for P10 under the same conditions) (Fig. 3b). An apparent quantum yield (AQY) of 3.02% was obtained at long wavelength of 595 nm (Fig. 3c), which places these A1/D1 DANHs among the most efficient photocatalysts for sacrificial hydrogen evolution in suspension-based systems (Table S1, ESI†). The AQYs recorded at 420, 490 and 515 nm were 3.72%, 3.43%, and 3.16%, respectively (Fig. 3c). These three similar AQY yields are consistent with the relatively flat UV-vis spectra of the sample in this spectral range (Fig. 3c).
Phase separation from solution was observed after photocatalysis (Fig. S8a and b, ESI†), which might suggest that this is a primary cause for loss of HER over time. SEM characterizations of the A1/D1 samples before and after photocatalysis supported particle aggregation (Fig. S9, ESI†). The A1/D1 samples were collected after photocatalysis and redissolved into THF solvent for characterization by UV-vis and 1H NMR spectroscopy. No obvious changes occurred before and after photocatalysis (Fig. S10, ESI†). We therefore suggest that the observed rate loss for A1/D1 is due to the aggregation of the DANHs during photocatalysis, rather than chemical decomposition. In support of this interpretation, A4/D1 samples exhibited a HER of 104.4 mmol g−1 h−1 in the first hour (Fig. 3b) but experienced a great loss of photocatalytic activity during irradiation. We observed that the A4/D1 NADHs aggregated much more quickly than the A1/D1 samples (Fig. S8c and d, ESI†), perhaps explaining the more rapid deactivation. Note also that no such deactivation occurs over 8 hours for P10, which is not nanoparticulate.
Finally, we extended these binary donor–acceptor nanohybrids to ternary donorA–donorB–acceptor nanohybrids (donorA:donorB = 1:1), and evaluated the photocatalytic activities using our high throughput photocatalysis screening platform. Compared to A1/D1 and A1/D2 binary DANHs, A1/D1:D2 ternary DANHs did not show enhanced photocatalytic activity (Fig. S11a, ESI†). Ternary A1/D2:D3, A1/D2:D5, A1/D3:D4 and A1/D4:D5 also showed no improvement (Fig. S11b–h, ESI†). However, intriguingly, in the presence of both D3 and D5, the hydrogen production rate of A1/D3:D5 was significantly improved. A1/D3:D5 reached a HER of 87.1 mmol g−1 h−1, which is much higher than the rate obtained for binary A1/D3 (1.6 mmol g−1 h−1) or A1/D5 (5.4 mmol g−1 h−1) DANHs with the same compositions (Fig. 3d). Enhancement to the performance was also accomplished in A1/D2:D4 DANHs (Fig. S12, ESI†). This shows that ternary donorA–donorB–acceptor compositions may outperform binary systems for photocatalysis in the future, raising an interesting multicomponent optimization challenge.
To summarize, high-throughput screening was used to discover both binary and ternary DANHs with sacrificial hydrogen evolution rates that greatly outperform the constituent donors and acceptors. This illustrates that a key principle from the field of organic photovoltaics can be translated into direct photocatalysis using organic materials. Non-fullerene acceptors gave higher photocatalytic performance, which is again relates to recent progress in OPV. Our results imply that catalyst lifetime may be limited by colloidal stability, rather than chemical decomposition, at least for short irradiation times (<1 day), suggesting the potential to improve catalyst lifetimes in the future by creating more stable colloids.
While this manuscript was under review McCulloch and co-workers reported a similar approach in using conjugated polymer/molecular acceptor composites for photocatalytic hydrogen evolution, also giving very high photocatalytic activities.29
We thank the Leverhulme Trust for studentship via the Leverhulme Research Centre for Functional Materials Design, and Engineering and Physical Sciences Research Council (EPSRC) for financial support under Grant EP/N004884/1. We thank Ni Wang and Chengxi Zhao for helpful discussions.
Conflicts of interest
There are no conflicts to declare.References
- K. Fujishima and A. Honda, Nature, 1972, 238, 37–38 CrossRef PubMed.
- X. Chen, S. Shen, L. Guo and S. S. Mao, Chem. Rev., 2010, 110, 6503–6570 CrossRef CAS PubMed.
- A. Kudo and Y. Miseki, Chem. Soc. Rev., 2009, 38, 253–278 RSC.
- Y. Wang, A. Vogel, M. Sachs, R. S. Sprick, L. Wilbraham, S. J. A. Moniz, R. Godin, M. A. Zwijnenburg, J. R. Durrant, A. I. Cooper and J. Tang, Nat. Energy, 2019, 4, 746–760 CrossRef CAS.
- X. Wang, K. Maeda, A. Thomas, K. Takanabe, G. Xin, J. M. Carlsson, K. Domen and M. Antonietti, Nat. Mater., 2009, 8, 76–80 CrossRef CAS PubMed.
- L. Lin, Z. Yu and X. Wang, Angew. Chem., Int. Ed., 2019, 58, 6164–6175 Search PubMed.
- D. J. Martin, K. Qiu, S. A. Shevlin, A. D. Handoko, X. Chen, Z. Guo and J. Tang, Angew. Chem., Int. Ed., 2014, 53, 9240–9245 Search PubMed.
- B. C. M. Martindale, E. Joliat, C. Bachmann, R. Alberto and E. Reisner, Angew. Chem., Int. Ed., 2016, 55, 9402–9406 Search PubMed.
- B. C. M. Martindale, G. A. M. Hutton, C. A. Caputo and E. Reisner, J. Am. Chem. Soc., 2015, 137, 6018–6025 CrossRef CAS PubMed.
- Y. S. Kochergin, D. Schwarz, A. Acharjya, A. Ichangi, R. Kulkarni, P. Eliášová, J. Vacek, J. Schmidt, A. Thomas and M. J. Bojdys, Angew. Chem., Int. Ed., 2018, 57, 14188–14192 Search PubMed.
- Z. Wang, X. Yang, T. Yang, Y. Zhao, F. Wang, Y. Chen, J. H. Zeng, C. Yan, F. Huang and J. X. Jiang, ACS Catal., 2018, 8, 8590–8596 CrossRef CAS.
- A. I. Cooper, Adv. Mater., 2009, 21, 1291–1295 CrossRef CAS.
- K. Wang, L. M. Yang, X. Wang, L. Guo, G. Cheng, C. Zhang, S. Jin, B. Tan and A. Cooper, Angew. Chem., Int. Ed., 2017, 56, 14149–14153 Search PubMed.
- S. Kuecken, A. Acharjya, L. Zhi, M. Schwarze, R. Schomäcker and A. Thomas, Chem. Commun., 2017, 53, 5854–5857 RSC.
- V. S. Vyas, F. Haase, L. Stegbauer, G. Savasci, F. Podjaski, C. Ochsenfeld and B. V. Lotsch, Nat. Commun., 2015, 6, 1–9 Search PubMed.
- X. Wang, L. Chen, S. Y. Chong, M. A. Little, Y. Wu, W. H. Zhu, R. Clowes, Y. Yan, M. A. Zwijnenburg, R. S. Sprick and A. I. Cooper, Nat. Chem., 2018, 10, 1180–1189 CrossRef CAS PubMed.
- P. Pachfule, A. Acharjya, J. Roeser, T. Langenhahn, M. Schwarze, R. Schomäcker, A. Thomas and J. Schmidt, J. Am. Chem. Soc., 2018, 140, 1423–1427 CrossRef CAS PubMed.
- S. Bi, C. Yang, W. Zhang, J. Xu, L. Liu, D. Wu, X. Wang, Y. Han, Q. Liang and F. Zhang, Nat. Commun., 2019, 10, 1–10 CrossRef PubMed.
- M. C. Scharber and N. S. Sariciftci, Prog. Polym. Sci., 2013, 38, 1929–1940 CrossRef CAS PubMed.
- L. Wang, R. Fernández-Terán, L. Zhang, D. L. A. Fernandes, L. Tian, H. Chen and H. Tian, Angew. Chem., Int. Ed., 2016, 55, 12306–12310 Search PubMed.
- P. B. Pati, G. Damas, L. Tian, D. L. A. Fernandes, L. Zhang, I. B. Pehlivan, T. Edvinsson, C. M. Araujo and H. Tian, Energy Environ. Sci., 2017, 10, 1372–1376 RSC.
- P. J. Tseng, C. L. Chang, Y. H. Chan, L. Y. Ting, P. Y. Chen, C. H. Liao, M. L. Tsai and H. H. Chou, ACS Catal., 2018, 8, 7766–7772 CrossRef CAS.
- S. Schubert, J. T. Delaney and U. S. Schubert, Soft Matter, 2011, 7, 1581–1588 RSC.
- Y. Bai, L. Wilbraham, B. J. Slater, M. A. Zwijnenburg, R. S. Sprick and A. I. Cooper, J. Am. Chem. Soc., 2019, 141, 9063–9071 CrossRef CAS PubMed.
- T. Ameri, J. Min, N. Li, F. Machui, D. Baran, M. Forster, K. J. Schottler, D. Dolfen, U. Scherf and C. J. Brabec, Adv. Energy Mater., 2012, 2, 1198–1202 CrossRef CAS.
- W. Zhao, S. Li, H. Yao, S. Zhang, Y. Zhang, B. Yang and J. Hou, J. Am. Chem. Soc., 2017, 139, 7148–7151 CrossRef CAS PubMed.
- J. Hou, O. Inganas, R. H. Friend and F. Gao, Nat. Mater., 2018, 17, 119–128 CrossRef CAS PubMed.
- M. Sachs, R. S. Sprick, D. Pearce, S. A. J. Hillman, A. Monti, A. A. Y. Guilbert, N. J. Brownbill, S. Dimitrov, X. Shi, F. Blanc, M. A. Zwijnenburg, J. Nelson, J. R. Durrant and A. I. Cooper, Nat. Commun., 2018, 9, 1–11 CrossRef CAS PubMed.
- J. Kosco, M. Bidwell, H. Cha, T. Martin, C. T. Howells, M. Sachs, D. H. Anjum, S. Gonzalez Lopez, L. Zou, A. Wadsworth, W. Zhang, L. Zhang, J. Tellam, R. Sougrat, F. Laquai, D. M. DeLongchamp, J. R. Durrant and I. McCulloch, Nat. Mater. DOI:10.1038/s41563-019-0591-1.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0cc00740d |
| This journal is © The Royal Society of Chemistry 2020 |