
Verification of sortase for protein conjugation by single-molecule force spectroscopy and molecular dynamics simulations†
 *a
*a
Abstract
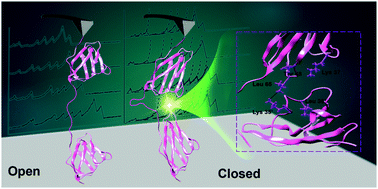
Sortase is one of the most widely used enzymes for covalent protein conjugation that links protein and protein/small molecules together in a site-specific way. It typically recognizes the “GGG” and “LPXTG” peptide sequences and conjugates them into an “LPXTGGG” linker. As a non-natural linker with several flexible glycine residues, it is unknown whether it affects the properties of the conjugated protein. To verify the use of sortase for protein–protein conjugation, we combined single-molecule force spectroscopy (SMFS) and molecular dynamics (MD) simulations to characterize sortase-conjugated polyprotein I27 with three different linkers. We found that the I27 with classic linkers “LPETGGG” and “LPETG” from sortase ligation were of normal stability. However, a protein with a longer artificial linker “LPETGGGG” showed a 15% lower unfolding force. MD simulations revealed that the 4G linker showed a high probability of a closed conformation, in which the adjacent monomer has transient protein–protein interaction. Thus, we verify the use of sortase for protein conjugation, and a longer linker with a higher glycine content should be used with caution.
 Please wait while we load your content...
Please wait while we load your content...

