
Significantly enhanced CO oxidation activity induced by a change in the CO adsorption site on Pd nanoparticles covered with metal–organic frameworks†
Hiroshi
Kitagawa  *aeh
*aeh
*aeh
Abstract
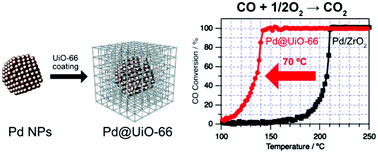
We report the significantly enhanced CO oxidation activity of Pd nanoparticles covered with [Zr6O4(OH)4(BDC)6] (UiO-66, BDC = 1,4-benzenedicarboxylate). The catalytic activity was much higher than those of Pd and Ru nanoparticles on ZrO2. The origin of the enhancement was suggested to be a change in the CO adsorption properties on Pd nanoparticles.
 Please wait while we load your content...
Please wait while we load your content...

