
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthesis, X-ray characterization and regium bonding interactions of a trichlorido(1-hexylcytosine)gold(III) complex†
Angel
Terrón
 *a,
Jordi
Buils
a,
Tiddo J.
Mooibroek
*b,
Miquel
Barceló-Oliver
a,
Angel
García-Raso
a,
Juan J.
Fiol
a and
Antonio
Frontera
*a
*a,
Jordi
Buils
a,
Tiddo J.
Mooibroek
*b,
Miquel
Barceló-Oliver
a,
Angel
García-Raso
a,
Juan J.
Fiol
a and
Antonio
Frontera
*a
aDepartment of Chemistry, Universitat de les Illes Balears, Crts de Valldemossa km 7.6, 07122 Palma de Mallorca (Baleares), Spain. E-mail: toni.frontera@uib.es; angel.terron@uib.es
bvan‘t Hoff Institute for Molecular Sciences, Universiteit van Amsterdam Science Park 904, 1098 XH Amsterdam, The Netherlands. E-mail: t.j.mooibroek@uva.nl
First published on 17th February 2020
Abstract
Herein we report the synthesis and X-ray characterization of a gold(III) complex of 1-hexylcytosine via N(3). The AuCl3N complexes stack on top of each other by reciprocal [Au⋯Cl] regium bonding interactions. After the first example 35 years ago, this is the second available structure of a cytosine nucleobase model complexed to gold(III).
One area of continuous interest is the interaction of nucleobases with transition metals due to their applications in the design of nanomaterials, MOFs and pharmacological principles.1–7 In particular, gold(III) complexes are drawing substantial attention due to their possible utilization in medicinal chemistry as antiparasitic, antibacterial, antiviral and antitumor agents.8–11 The most representative types of bioactive gold(III) compounds are dithiocarbamates.12 N-heterocycles,13 porphyrinato complexes14 and dinuclear oxo-complexes.15
Pure gold(III)-derivatives are difficult to obtain due to the fact that Au(III) ion has high reduction potential. Several strategies have been used in the literature to synthesize physiologically stable9 gold(III) complexes, which are mostly related to the selection of the appropriate ligand to decrease the strong propensity of the Au(III) ion to be reduced to Au(I)/Au(0). This is the reason behind the scarce number of Au(III) nucleobase complexes available in the literature. As a matter of fact, Lippert and Freisinger16 have demonstrated the oxidation of guanine ring when it is bound to Au(III), in line with other studies that evidenced the degradation of Au(III)-(ethylendiamine)-(guanosine 5′-monophosphate) complexes.17 Additional problems to obtain stable gold(III) complexes arise from their sensitivity to light.18
It should be emphasized that only twelve X-ray structures between purine or pyrimidine(-like) bases and gold(III) have been described so far (see Fig. S1, ESI†). Among them, the more relevant are: (i) Au(III)-(1,9-dimethylguanine)16 where gold is coordinated through N(7); (ii) Au(III) bonded to the C(5) atom of 1,3-dimethyluracilato19 and (iii) Au(III) coordinated to N(3) of a 1-methylcytosine.20 Moreover, a pair of bases linked by gold (cytidine–Au–guanine) has been also described;21 however the resolution was too low and, consequently, neither the oxidation state of gold, nor the coordination points of the metal were resolved. Curiously, only one cytosine derivative complexed to gold(III) is known, which was reported 35 years ago by Wong's group.20 Thus, we present herein the synthesis and X-ray characterization of the second structure of gold(III) complexed to a cytosine nucleobase model (N1-hexylcytosine) (compound 1 see Scheme 1) thus extending the short list of X-ray structurally characterized examples of gold(III)-cytosine model complexes. Moreover, we describe and analyse the Au⋯Cl noncovalent contacts observed in the solid state of compound 1 that can be classified as π-hole22 regium bonds23 as further discussed below.
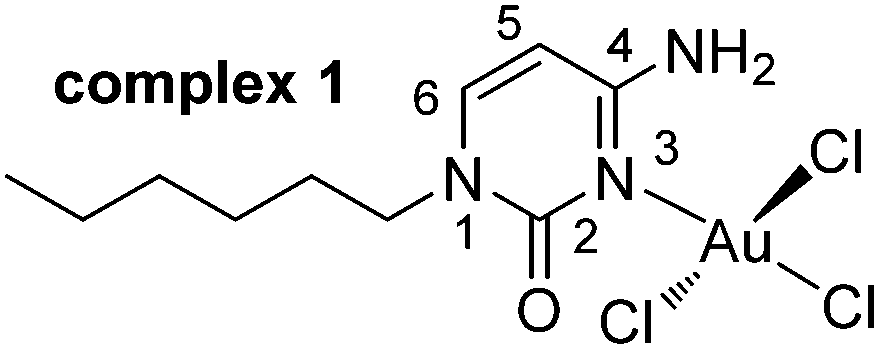 | ||
| Scheme 1 Gold(III) complex 1 with numbering of cytosine ring. | ||
Compound 1 was synthesized in 49% yield by mixing an equimolar amount of Na[AuCl4]·2H2O (in water) and N1-hexylcytosine (in dioxane) for one day in the absence of light (see ESI† for details). The X-ray structure of compound 1 along with the numbering scheme is shown in Fig. 1. The compound crystallizes in the monoclinic P21/c space group and the AuCl3 entity is coordinated to the N(3) nitrogen atom of cytosine ring. The geometry of the Au(III) ion is essentially square-planar and the coordination plane forms an angle of 74.35° with the cytosine ring.
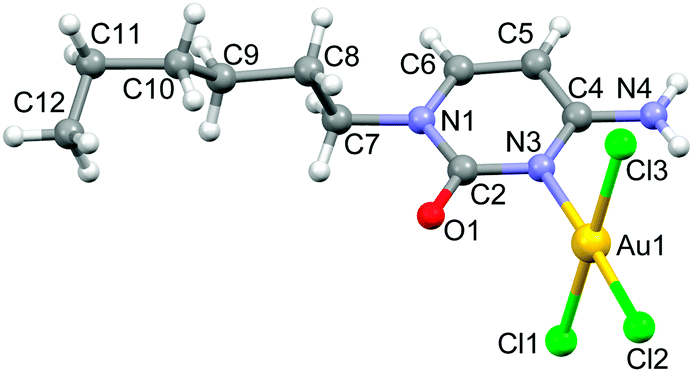 | ||
| Fig. 1 Ball & stick representation of the X-ray structure of compound 1 along with the atom numbering scheme of non-hydrogen atoms. | ||
In the solid state compound 1 forms 2D layers (see Fig. 2, top) where molecules self-assemble via N–H⋯Cl and Au⋯Cl contacts to form dimers that further interact to each other by hydrophobic C–H⋯H–C interactions (highlighted in light green) involving the hexyl chains. The hydrogen bonding interactions can be considered as moderate from a geometric point of view, as they are characterized by H⋯Cl distances of 2.661 Å and N–H⋯Cl angles of 153.22° (see Fig. S3 for details, ESI†). More interesting are the Au⋯Cl distances of 3.545(3) Å and the accompanying Au–Cl⋯Au angles of 88.17° (see Fig. 2, bottom). The intermolecular distance is significantly longer than the sum of Au and Cl covalent radii (i.e. 2.38 Å) and slightly longer than the sum of van der Waals radii (3.41 Å) if Bondi's value (1.66 Å) is used for gold.24 However, in 2001 Batsanov25 demonstrated that this value is underestimated and suggested a better radius of 1.86 Å for gold. Earlier investigations proposed even larger values for gold's radius (RvdW > 2 Å).26 Therefore, if the Batsanov's value is used the Au⋯Cl contact in compound 1 becomes slightly shorter than ∑RvdW (3.61 Å).
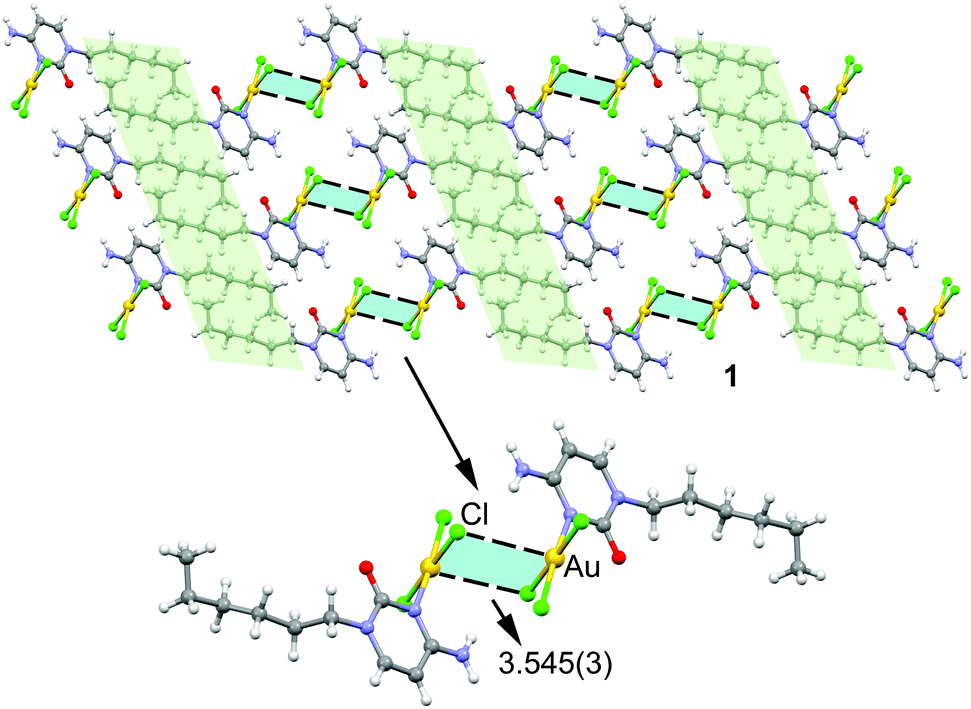 | ||
| Fig. 2 Partial view of the crystal packing of compound 1 with indication of the regium bonds. Distance in Å. | ||
We have theoretically analysed the Au⋯Cl interaction in 1. In this context, a recent investigation27 introduced the concept “regium bond” to describe the noncovalent interaction between a noble metal acting as a Lewis acid and any electron rich moiety, which was contextualized as example of σ-hole bonding. In analogy to σ-hole (a region of electropositive potential located on the extension of a σ-bond), a π-hole is defined as a region of electropositive potential located in the direction perpendicular to a σ-framework of the molecular entity. To verify the existence of a π-hole in a gold(III) AuCl3N fragment, we have computed the molecular electrostatic potential (MEP) surface, which is shown in Fig. 3. The MEP surface clearly evidences the existence of a region of positive potential above and below the Au atom (+11 kcal mol−1). The maximum MEP value is located on the NH2 H-atoms (+25 kcal mol−1). The most negative MEP is located at the AuCl3N plane between two chlorido ligands (−41 kcal mol−1). It is interesting to note that the MEP value at the belts of the Cl atoms are negative thus suitable to participate in regium bonding interaction as π-hole acceptors (Lewis base).
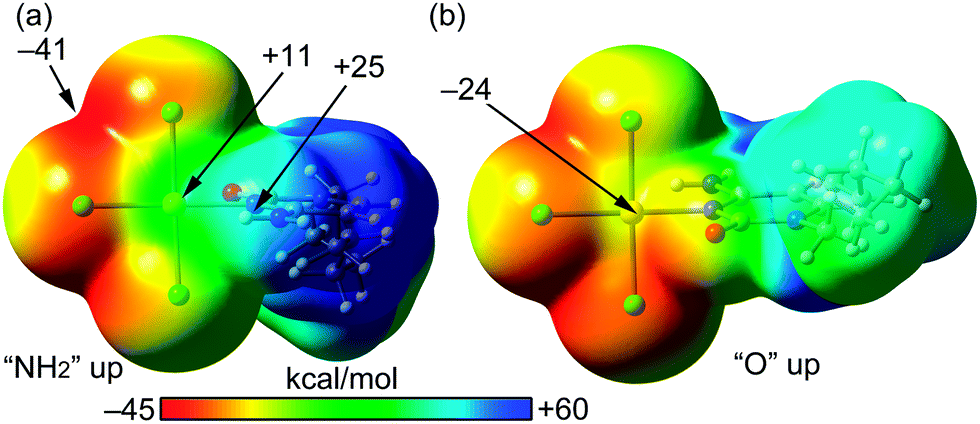 | ||
| Fig. 3 MEP surfaces (0.001 a.u.) of complex 1 at the PBE0-D3/def2-TZVP level of theory, using two different “on-top” orientations (a and b). The energies at selected points are given in kcal mol−1. | ||
We have examined and characterized the self-assembled dimer retrieved from the solid state of 1 (see Fig. 2, bottom) by using DFT (PBE0-D3/def2-TZVP) calculations (see ESI† for details). Fig. 4 shows that in addition to the formation of the regium bonds, two symmetrically equivalent H-bonds are also established between the exocyclic amino group and the chlorido ligand (red dashed lines, H-bond distance 2.66 Å). The dimerization energy is large and negative (ΔE1 = −21.6 kcal mol−1) due to the formation of two H-bonds and two regium bonding interactions simultaneously. In an effort to roughly estimate the contribution of the regium bonds, we have used a theoretical model where the amino groups have been replaced by H-atoms (see small red arrows in Fig. 4b). Consequently, the H-bonds cannot be formed and the interaction energy is reduced to ΔE2 = −15.9 kcal mol−1 (around −8 kcal mol−1 each regium bond). Therefore, the contribution of the regium bonds is more important than that of H-bonds, thus emphasizing the importance of the Au⋯Cl contacts in the dimerization process. Interestingly, a dimer where the one regium bond is ‘broken’ by forcing one Au–Cl⋯Au angle near 180° was computed to give −6.2 kcal mol−1 stabilization energy (see Fig. S2, ESI†). This is less than half of ΔE2, which can be understood by the lower electron density at the extension of an Au–Cl vector (see also Fig. 3) and/or by a synergy of the reciprocal interactions as both monomers act simultaneously as electron donor (Cl) and acceptor (Au).
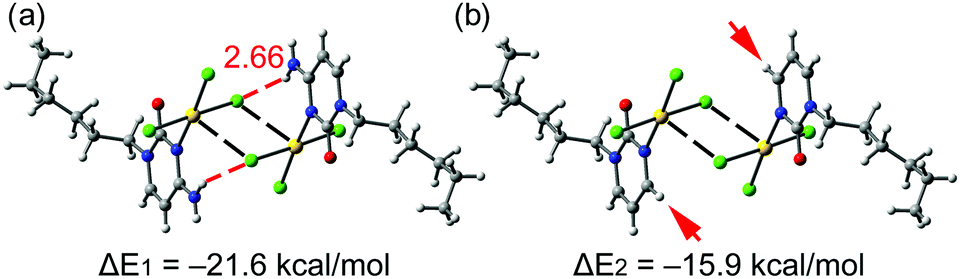 | ||
| Fig. 4 Energetic features at the PBE0-D3/def2-TZVP level of theory of the self-assembled dimer of 1 (a) and a theoretical dimer where the exocyclic amino groups have been replaced by H-atoms (b). | ||
To further evidence the existence of the regium bonds, we have used two additional computational tools. Bader's quantum theory of “atoms in molecules” (QTAIM)28 and the noncovalent interaction plot (NCIPlot) index.29 The existence of a bond critical point (CP) and a bond path connecting to atoms derived from the QTAIM theory is an unambiguous evidence of an interaction.30 The NCIPLOT is a computational index that is convenient for the easy visualization and identification of NCIs. The noncovalent contacts are identified with the peaks that emerge in the reduced density gradient (RDG) at low densities.31 These are plotted by mapping an isosurface of s (s = |∇ρ|/ρ4/3) for a low value of RDG. When a supramolecular dimer is formed, the RDG changes at the CPs in between the monomers due to the annihilation of the density gradient at these CPs. Therefore, this index visualizes the extent to which NCIs stabilize a supramolecular assembly qualitatively and reveals which molecular regions interact. The color code is red-yellow-green-blue where the red and yellow colors are used for strong and weak repulsive (ρcut+), respectively and the blue and green colors for strong and weak attractive (ρcut−) forces, respectively. The distribution of bond CPs and bond paths is given in Fig. 5a.
 | ||
| Fig. 5 (a) AIM distribution of bond and ring CPS (red and yellow spheres) and bond paths of the dimer of compound 1. The values of charge density (ρ), potential energy density (V) and Lagrangian kinetic energy density (G) are given in a.u. (b) NCIPlot of the dimer of compound 1. The parameters used to build the surface are: gradient cut-off = 0.35 a.u. and color scale −0.04 < ρ < 0.04 a.u. | ||
The presence of two symmetrically equivalent bond CPs and bond paths inter-connecting the Cl and Au atoms can clearly be observed, thus confirming the existence of the interaction. Moreover, the distribution also reveals the existence of the two symmetrically equivalent H-bonds that are characterized by a bond CP and bond path connecting the H-atom to the chlorido ligand. The values of density, potential energy density and Lagrangian kinetic energy density [ρ(r), V(r) and K(r) in a.u., respectively] at the CPs are also indicated in Fig. 5a, which are comparable for both interactions and they are in the typical range of noncovalent forces.28 The V(r) is greater in absolute value for the regium bond than the H-bond, which agrees with the fact that the regium bond is stronger than the H-bond. The NCIPlot is represented in Fig. 5b and provides a further evidence of the attractive nature of the Au⋯Cl regium bond. The isosurfaces that characterize the regium bonds are bigger and also embraces the Au–Cl covalent bonds, resembling antiparallel CO⋯CO dipole⋯dipole interactions. The additional complementarity between the Au–Cl bonds may explain the stronger nature of the regium bonds compared to H-bonds.
We have also studied if orbital contributions are important in the π-hole regium bonding interactions described above. For this purpose, we have carried out natural bond orbital (NBO) calculations focusing on the second order perturbation analysis that is adequate to analyse donor–acceptor interactions.32 Remarkably this analysis reveals a significant orbital contribution that comes from an electron donation (LP → p) from the one lone pair (LP) at the chlorine atom to an empty LP orbital of Au (basically constituted by the 6pz atomic orbital), with a concomitant stabilization energy of 10.1 kcal mol−1. This result confirms the π-hole nature of the interaction.
Finally, we have analysed the Cambridge Structure Database to investigate if these type of regium bonds in tetra coordinated gold complexes are common and potentially important in crystal engineering (see ESI† for details). Shown in the left-hand side of Fig. 6 is the heat plot of d′ versus α for a query where Y is an electron rich atom (ElR) and the (intermolecular) cut-off distance d is the sum of the Bondi van der Waals radii24 of Au and ElR + 2 Å.
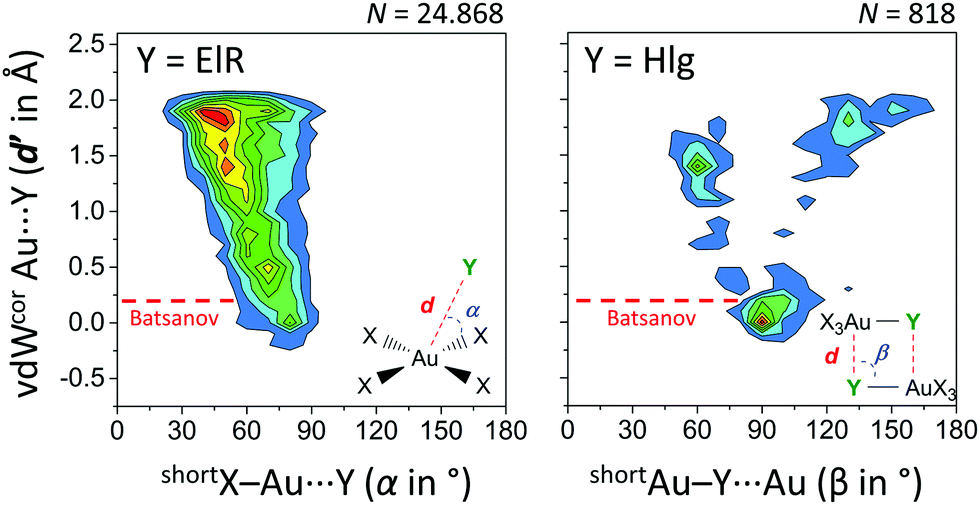 | ||
| Fig. 6 Heat plots of van der Waals corrected intermolecular Au⋯Y distances (d′, derived from d and the Bondi van der Waals radii of Au and Y)24versus angle α or β (the shortest was used). Left: Y = any electron rich atom (N, P, As, O, S, Se, Te, F, Cl, Br, I, At) within 2.781 CIFs. Right: Y = any halogen (‘Hlg’; F, Cl, Br, I, At) within 523 CIFs. X = any atom and both queries (illustrated in the inset figures) involved data with Au⋯Y distances d′ ≤ 2 Å. Color scales increase from blue to red and are up to 2.5% (left) or 3.5% (right). The red line indicates van der Waals overlap using Batsanov's van der Waals radius for Au (1.86 Å).25 | ||
Most of these data are above the Bondi and Batsanov van der Waals benchmark, although a small grouping of data is noticeable around d′ = 0 Å and α = 80°. This feature is more pronounced for data involving [AuX4]⋯Hlg (Fig. S1, Hlg = any halogen atom, ESI†) and most pronounced in data with [AuHlg3N]⋯ElR (Fig. S2, where Hlg = Cl in 93% of cases, ESI†). The heat plot of a search involving X3Au–Hlg dimers is shown in the right-hand side of Fig. 6. About 31% of these data is grouped around β = 90° with d′ ≤ 0.5 Å. Interestingly, nearly all of these data display van der Waals overlap using Batsanov's van der Waals radius for Au (1.86 Å). Moreover, the grouping at β = 90° corresponds well with the observed Au–Cl⋯Au angles of 88.17° in the crystal structure of 1. Both plots in Fig. 6 thus reveal the geometric preference that shorter intermolecular Au⋯Y distances (d′) correlate with X–Au⋯Y angles nearer 90°.
In conclusion, herein we have reported the synthesis and X-ray characterization of the second known cytosine-derived gold(III) complex (1). Structure 1 exhibits previously overlooked Au⋯Cl regium bonding interactions, as verified by various DFT calculations. The LP → p nature of the interaction confirms that it can be classified as a π-hole regium bond. To our knowledge this is the first report on regium bonding involving gold(III). Finally, the results from the CSD search confirm that this type of bonds are common in X-ray structures and have remained basically unnoticed likely due to the underestimated van der Waals radius value that is tabulated for gold.
We thank the MINECO/AEI from Spain for financial support (project numbers CTQ2017-85821-R and CTQ2017-90802-REDT, FEDER funds). We thank the Vice-Rector for Research and International Relations of the UIB for the financial support in setting up the single-crystal X-ray diffraction facility. TJM thanks NWO for funding (VIDI project 723.015.006).
Conflicts of interest
There are no conflicts to declare.Notes and references
- P. Zhou, R. Shi, J. F. Yao, C. F. Sheng and H. Li, Coord. Chem. Rev., 2015, 292, 107 CrossRef CAS.
- F. Pu, J. Ren and X. Qu, Chem. Soc. Rev., 2018, 47, 1285 RSC.
- Handbook of nucleobase complexes, Transition Metal complexes of naturally occurring nucleobases and their derivatives, ed. R. Lusty, P. Wearden and V. Moreno, CRC Press, Boca Ratón, 1990 and 1992, vol I and II Search PubMed.
- B. Lippert and P. J. San Miguel, Acc. Chem. Res., 2016, 49, 1537 CrossRef CAS PubMed.
- B. Lippert and P. J. San Miguel, Adv. Inorg. Chem., 2018, 71, 278 CrossRef.
- Interaction of metal ions with nucleotides, nucleic acids, and their constituents, Metal Ions in Biological Systems, ed. A. Sigel and H. Sigel, 1996, vol. 32 Search PubMed.
- Interplay between Metal Ions and Nucleic Acids, Metal Ions in Life Sciences, ed. A. Sigel, H. Sigel and R. K. O. Sigel, Springer, 2012, vol. 10 Search PubMed.
- B. Đ. Glišić, N. D. Savić, B. Warżajtis, L. Djokic, T. Ilic-Tomic, M. A. Slavko Radenkovic, J. Nikodinovic-Runic, U. Rychlewska and M. I. Djurana, Med. Chem. Commun., 2016, 7, 1356 RSC.
- S. J. Berners-Price, Gold-based therapeutic agents: a new perspective, in Bioinorganic Medicinal Chemistry, ed. E. Alessio, Wiley-VCH Verlag GmbH & Co. KGaA, Wienheim, 2011, p. 197 Search PubMed.
- B. Bertrand and A. Casini, Dalton Trans., 2014, 43, 4209 RSC.
- B. Đ. Glišić and M. I. Djuran, Dalton Trans., 2014, 43, 5950 RSC.
- D. Saggioro, M. P. Riogobello, L. Paloschi, A. Folda, S. A. Moggach, S. Parsons, L. Ronconi, D. Fregona and A. Bindoli, Chem. Biol., 2007, 14, 1128 CrossRef CAS PubMed.
- C. K.-L. Li, R. W.-Y. Sun, S. C.-F. Kui, N. Zhu and C.-M. Che, Chem. – Eur. J., 2006, 12, 5253 CrossRef CAS PubMed.
- R. W.-Y. Sun and C.-M. Che, Coord. Chem. Rev., 2009, 253, 1682 CrossRef CAS.
- (a) A. Casini, M. A. Cinellu, G. Minghetti, C. Gabbiani, M. Coronnello, E. Mini and L. Messori, J. Med. Chem., 2006, 49, 5524 CrossRef CAS PubMed; (b) M. A. Cinellu, L. Maiore, M. Manassero, A. Casini, M. Arca, H.-H. Fiebig, G. Kelter, E. Michelucci, G. Pieraccini, C. Gabbiani and L. Messori, ACS Med. Chem. Lett., 2010, 1, 336 CrossRef CAS PubMed.
- A. Schimanski, E. Freisinger, A. Erxleben and B. Lippert, Inorg. Chim. Acta, 1998, 283, 223 CrossRef CAS.
- S. Zhu, W. Gorski, D. R. Powell and J. A. Walmsley, Inorg. Chem., 2006, 45, 2688 CrossRef CAS PubMed.
- L. A. Espinosa-Leal and O. Lopez-Acebedo, Nanotechnol. Rev., 2015, 4, 173 CAS.
- F. Zamora, E. Zangrando, M. Furlan, L. Randaccio and B. Lippert, J. Organomet. Chem., 1998, 552, 127 CrossRef CAS.
- M. S. Holowczak, M. D. Tanci and G. S. Wong, J. Am. Chem. Soc., 1985, 107, 5789 CrossRef CAS.
- E. Ennifar, P. Walter and P. Dumas, Nucleic Acids Res., 2003, 31, 2671 CrossRef CAS PubMed.
- (a) A. Bauzá, T. J. Mooibroek and A. Frontera, ChemPhysChem, 2015, 16, 2496 CrossRef PubMed; (b) A. Bauzá and A. Frontera, Inorganics, 2018, 6, 64 CrossRef; (c) A. Frontera and A. Bauzá, Chem. – Eur. J., 2018, 24, 7228 CrossRef CAS PubMed.
- (a) J. H. Stenlid and T. Brinck, J. Am. Chem. Soc., 2017, 139, 11012 CrossRef CAS PubMed; (b) G. Sanchez-Sanz, C. Trujillo, I. Alkorta and J. Elguero, ChemPhysChem, 2019, 20, 1572 CrossRef CAS PubMed; (c) B. Zheng, Y. Liu, Z. Wang, F. Zhou, Y. Liu, X. Ding and T. Lu, Mol. Phys., 2019, 117, 2443 CrossRef CAS; (d) W. Zierkiewicz, M. Michalczyk and S. Scheiner, Phys. Chem. Chem. Phys., 2018, 20, 22498 RSC; (e) J. Halldin Stenlid, A. J. Johansson and T. Brinck, Phys. Chem. Chem. Phys., 2018, 20, 2676 RSC.
- A. Bondi, J. Phys. Chem., 1964, 68, 441 CrossRef CAS.
- S. S. Batsanov, Inorg. Mater., 2001, 37, 871 CrossRef CAS.
- (a) S. S. Batsanov, Zh. Fiz. Khim., 2000, 74, 1273 CAS; (b) S. S. Batsanov, J. Mol. Struct., 1999, 468, 151 CrossRef CAS; (c) S. S. Batsanov, Zh. Neorg. Khim., 1991, 36, 3015 CAS.
- J. H. Stenlid and T. Brinck, J. Am. Chem. Soc., 2017, 139, 11012 CrossRef CAS PubMed.
- R. F. W. Bader, Chem. Rev., 1991, 91, 893 CrossRef CAS.
- J. Contreras-García, E. R. Johnson, S. Keinan, R. Chaudret, J.-P. Piquemal, D. N. Beratan and W. Yang, J. Chem. Theory Comput., 2011, 7, 625 CrossRef PubMed.
- R. F. W. Bader, J. Phys. Chem. A, 1998, 102, 7314 CrossRef CAS.
- E. R. Johnson, S. Keinan, P. Mori-Sanchez, J. Contreras-Garcia, A. J. Cohen and W. Yang, J. Am. Chem. Soc., 2010, 132, 6498 CrossRef CAS PubMed.
- F. Weinhold and C. R. Landis, Valency and Bonding: A Natural Bond Orbital Donor-Acceptor Perspective, Cambridge University Press, Cambridge, UK, 2005 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details, theoretical methods and crystallographic table. CCDC 1978213. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0cc00505c |
| This journal is © The Royal Society of Chemistry 2020 |