DOI:
10.1039/D0CC00482K
(Communication)
Chem. Commun., 2020,
56, 3365-3368
Key factor for the anti-Arrhenius low-temperature heterogeneous catalysis induced by H+ migration: H+ coverage over support†
Received
18th January 2020
, Accepted 25th February 2020
First published on 13th March 2020
Abstract
Low-temperature heterogeneous catalytic reaction in an electric field is anticipated as a novel approach for on-demand and small-scale catalytic processes. This report quantitatively reveals the important role of proton coverage on the catalyst support for catalytic ammonia synthesis in an electric field, which shows an anti-Arrhenius behaviour.
During the past several decades, industrial catalytic processes have been optimized for large-scale processes. They are operated efficiently under harsh conditions (high temperatures and pressures) using various facilities such as heat exchangers. However, for next-generation catalytic processes supporting a sustainable society, small-scale plants that work under milder conditions are anticipated for on-site and on-demand operation. To moderate catalyst working conditions, catalytic reactions using external stimuli (e.g. photonic, magnetic, and electric fields) have attracted great attention.1–8 Our group, which has specifically examined the use of an electric field for low-temperature catalysis, has facilitated various catalytic reactions such as steam reforming of hydrocarbon, dehydrogenation of methylcyclohexane (MCH), and NH3 synthesis.9–23 In an electric field, H+ hopping via surface hydroxyl groups proceeds (so-called surface protonics), thereby enabling the reaction path through collision between H+ over support and adsorbent over loading metals such as CH4, MCH, and N2.
Various analyses have revealed novel catalysis qualitatively in an electric field induced by H+ hopping.9–23 Quantitative evaluation of the H+ contribution to the overall reaction rate is extremely important. The work described herein succeeded in formulizing the effect of H+ amount over supports in the electric field using NH3 synthesis and Ru/CeO2 respectively as a model reaction and catalyst.
For this study, 1 wt%Ru/CeO2 was prepared using an impregnation method with CeO2 (JRC-CEO-01) as a support. The effects on the NH3 synthesis rate (r) were observed when using an electric field (6 mA direct current) and H2 partial pressure (PH2) at various catalyst bed temperatures. Before all activity tests, the catalyst was pre-reduced under a common condition (N2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :H2 = 1:3 (PH2 = 0.75 atm), 723 K, 2 h). A schematic image of the fixed-bed type reactor is presented in Fig. S1 (ESI†). The catalyst bed temperature was detected directly using a thermocouple attached on the catalyst particles to exclude Joule heat effects on the reaction caused by the imposed current.
:H2 = 1:3 (PH2 = 0.75 atm), 723 K, 2 h). A schematic image of the fixed-bed type reactor is presented in Fig. S1 (ESI†). The catalyst bed temperature was detected directly using a thermocouple attached on the catalyst particles to exclude Joule heat effects on the reaction caused by the imposed current.
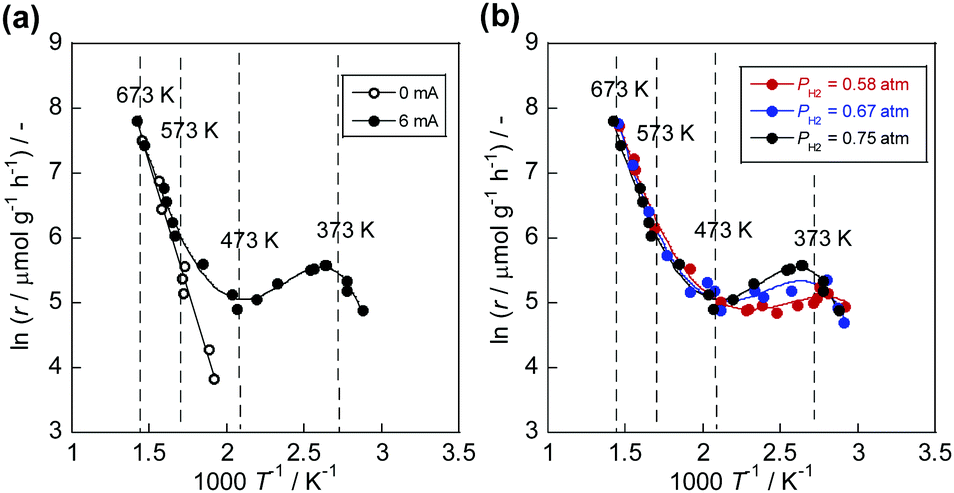
Without the electric field, a conventional-shaped Arrhenius plot was obtained (open symbols in Fig. 1(a)). In contrast, the electric field induced completely different temperature dependence of the NH3 synthesis rate (closed symbols in Fig. 1(a) and Tables S1, S2, ESI†). Conventional Arrhenius-like behaviour was detected in the high-temperature region (573–673 K). However, the slope decreased with the temperature decrement. Also, anti-Arrhenius-like behaviour emerged around 373–573 K. Subsequently, Arrhenius-like behaviour was confirmed again at temperatures lower than around 373 K. This peculiar trend suggests a change in the reaction mechanism depending on the catalyst bed temperature. The PH2 dependence supported this assumption (Fig. 1(b) and Tables S3–S6, ESI†). At around 373–573 K, at which anti-Arrhenius-like behaviour was observed, PH2 has a positive effect on the reaction rate, although the reaction rates changed slightly at other temperature regions. Conventionally, Ru-based catalysts show negative dependence on PH2 because of H2 poisoning over the Ru surface.24–26 When the CeO2 is used as the support, the poisoning is suppressed by virtue of the spillover of H atom from Ru surface to CeO2.27 Without the electric field, we also confirmed a slight change of activities with increasing PH2 (Fig. S2, ESI†). Therefore, enhancement of the activities by high PH2 over Ru/CeO2 is a completely specific trend in the electric field, indicating a novel reaction mechanism.
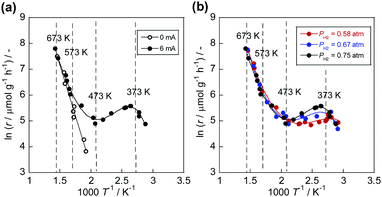 |
| | Fig. 1 Arrhenius plots for NH3 synthesis rate (r) over 1 wt%Ru/CeO2: (a) with/without the electric field (0 or 6 mA) under PH2 = 0.75 atm; and (b) PH2 dependence in the electric field (0.58, 0.67, and 0.75 atm) with 6 mA. | |
Reportedly, conventional ammonia synthesis proceeds via the ‘dissociative mechanism’, where supplied N2 directly dissociates.28
However, most recently, we have inferred that ammonia synthesis in the electric field proceeds through the ‘associative mechanism’, where N
2 associates with H
+ over supports before dissociation.
19| | | N2ad + H+ad + e− → N2Had | (6) |
| | | N2Had + H+ad + e− → N2Had | (7) |
| | | N2H2ad + H+ad + e− → Nad + NH3(g) | (8) |
Here, ‘ad’ denotes adsorbed species, and ‘
g’ means gaseous species. In addition, N
xH
y (
x = 0–2,
y = 0–2) adsorbs over loaded metals and H
+ adsorbs over lattice oxygen of supports (O
lat). Similarly, we have reported C–H cleavage activation by hopping proton over a support.
9–18 Therefore, the amount of H
+ over the support is expected to be crucially important for novel catalysis in the electric field. We assumed, therefore, that the temperature dependence of the H
+ coverage over CeO
2 plays an important role in specific temperature dependence with the electric field.
To elucidate this hypothesis quantitatively, the temperature dependence of H+ coverage over Olat under PH2 = 0.75 atm was investigated using in situ FT-IR measurements in a transmission mode (Fig. 2(a)) without the electric field and DRIFTs mode (Fig. 2(b)) with/without the electric field. The hydrogen adsorption energy over Olat (H+ stability over Olat) was fundamentally important for the NH3 synthesis rate in the electric field.19 To exclude effects of atmospheric H2O outside of the measurement cell, D2 was supplied as a reactant. Also, Olat-D+ stretching peaks (around 2780 cm−1)29 were used for pseudo-quantitative analysis of the H+ amount at each temperature. Detailed measurement procedures are shown in Fig. S3 (ESI†). The IR cell for transmission mode is suitable for a quantitative investigation of surface OH group (Fig. 2(c)), on the other hand the electric field is not applicable for the cell due to its structure. So we also measured DRIFTs for evaluating the effect of the electric field on the amount of surface OH group (note that DRIFTs cannot evaluate the precise quantitative amount due to its principle). The qualitative analysis with the electric field (Fig. 2(b)) showed that no significant change was observed by the application of electric field. Electric field application produces small amount of Joule heat, so small change can be observed, but the effect of gas phase temperature (i.e. difference in between 323 K and 473 K) on the adsorption is much larger.
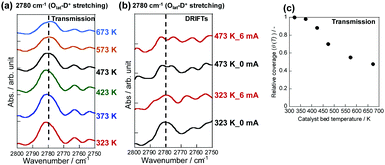 |
| | Fig. 2 Quantitative analysis for Olat-D+ over 1 wt%Ru/CeO2 under N2 5 SCCM and D2 15 SCCM: (a) in situ IR transmission spectra, (b) in situ IR DRIFTs spectra with/without EF, and (c) relative coverage by transmission spectra (θ(T) = Area(T)/Area(323 K)). | |
We calculated the relative coverage of H+ (θ(T)) using the peak area, where the value at 323 K (Area (323 K)) was used as a reference (eqn (9)).
| | | θ(T) = Area(T)/Area(323 K) | (9) |
Results revealed that the H
+ coverage decreased with the increment in temperatures (
Fig. 2(c)). Furthermore, at 323–373 K, the H
+ coverage changed slightly, indicating saturation of H
+ coverage.
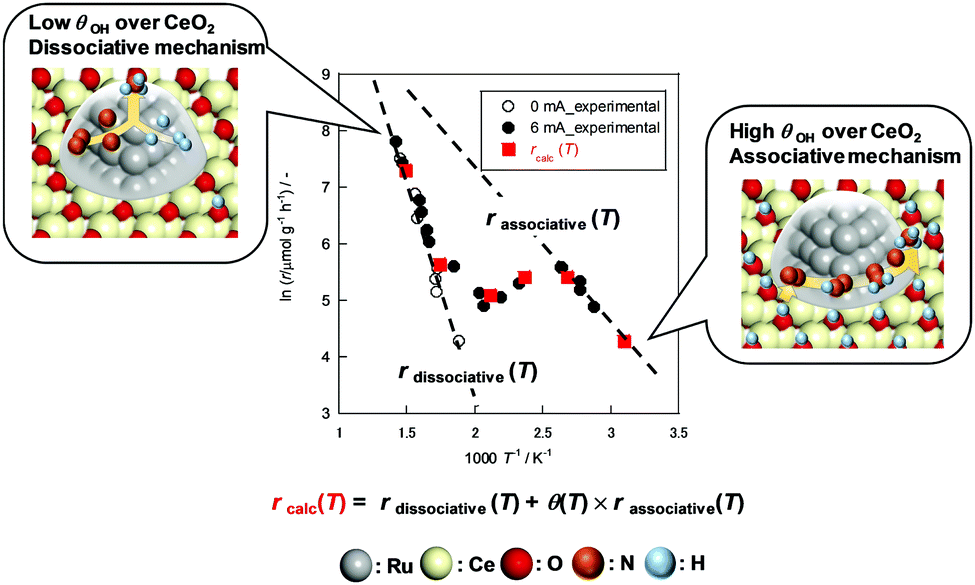
The obtained θ(T) suggested the contribution of H+ coverage dependence on the temperature to the specific Arrhenius plots in the electric field. When catalyst bed temperatures are higher than 573 K, the decrement in H+ over CeO2 renders the NH3 synthesis rate acceleration negligible, which means that NH3 synthesis at the high-temperature region proceeds via a conventional ‘dissociative mechanism’. Fig. 2 reveals that the Olat-D+ remained on the surface to some extent even at high temperature region, however, the influence became small from following reasons. At first, the reaction through ‘dissociative mechanism’ is much active at high temperature region. Furthermore, the ‘associative mechanism’ is limited at the three phases boundary (TPB),22 although the ‘dissociative mechanism’ can proceed over all region of metal surface. It means that the reaction site for ‘dissociative mechanism’ is much larger than that for ‘associative mechanism’. Therefore, the contribution of ‘associative mechanism’ to the overall reaction rate became very small at high temperature region even if there is proton over supports. In contrast, the CeO2 surface is covered by H+ sufficiently at temperatures lower than 373 K; also, H+ reacts with N2 over Ru. The overall reaction rate at around 373–573 K can be described as the sum of two rates of reaction through the ‘dissociative mechanism’ and the ‘associative mechanism.’ Therefore, we assumed that the overall NH3 synthesis rate in the electric field is definable as
| | | rcalc(T) = rdissociative(T) + θ(T) × rassociative(T) | (10) |
where
rdissociative(
T) and
rassociative(
T) show the extrapolated values (
Fig. 3). The Arrhenius plots without the electric field (
Fig. 1(a)) are used for
rdissociative(
T).
rassociative(
T) was calculated using approximate straight line which was drawn using experimental values at low-temperature (
T < 373 K) region. At the region, negligible change of
θ(
T) was detected, and Arrhenius-like behaviour was confirmed for NH
3 synthesis rate in the electric field. Therefore, we concluded that the straight line at right side in
Fig. 3 would mean the ideal NH
3 synthesis rate through ‘associative mechanism’ with maximum H
+ coverage. Rate determining step (RDS) approximation was used for the second term of the
eqn (10). We inferred that the RDS in the electric field might be the N
2H formation step (
eqn (6)), meaning that the reaction rate would be scaled linearly by the H
+ concentration.
19 As for the H
+ concentration, the actual measured values presented in
Fig. 2(c) are used.
Fig. 3 shows a comparison between
rcalc(
T) and the experimental value. For this consideration,
r under
PH2 = 0.75 atm with 6 mA was used. It is particularly interesting that the calculated value showed close agreement with the experimental value. The coincidence reveals the linear correlation between the enhancement of the reaction rate and H
+ amount in the electric field. It also supports the credibility of RDS approximation on N
2H formation step. This result also corresponds to the
PH2 dependence shown in
Fig. 1(b). The temperature of inflection points around 373 K became lower. Under
PH2 = 0.78 atm, the H
+ coverage hits ceiling at around 373 K, and the conventional temperature dependence emerged. When the
PH2 decreased, the temperature at which reaches a limit became lower owing to the decrease of H
+ source.
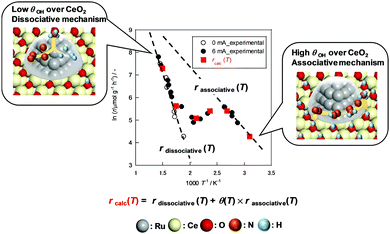 |
| | Fig. 3 Experimental reaction rate under PH2 = 0.78 atm and calculated reaction rate (rcalc(T)). Data in Fig. 1(a) are used for rdissociative and rassociative. | |
This report is the first study that quantitatively assesses the relation between H+ coverage and reaction rates in the electric field. This finding is expected to be expanded to other reaction systems involving H+ and is expected to constitute an essential guideline for the optimization of catalyst and reaction conditions in an electric field. For instance, when the reaction was conducted in a low-PH2 atmosphere, catalysts should be designed to keep H+ tightly. In contrast, under high PH2, catalysts that bind H+ loosely would be demanded to enhance the H+ supply ability.19
In summary, we analysed the novel catalysis quantitatively and qualitatively under an electric field using NH3 synthesis as a model reaction. We identified a peculiar temperature dependence of the NH3 synthesis rate in the electric field. Three regions were confirmed: anti-Arrhenius-like behaviour emerged at around 373–573 K; Arrhenius-like behaviour was observed in the other two temperature regions (T > 573 K or T < 373 K). Furthermore, PH2 dependence on the electric field was influenced by the temperature. The activities increase with high PH2 only around 373–573 K. The H+ coverage was detected using in situ FT-IR measurements in a transmission mode. The results of analysis showed that the increment of H+ coverage accorded to the temperature decrease. The coverage hits a ceiling at around 373 K. Based on the findings described above, the overall reaction rate in the electric field was formulated. The calculated reaction rate fitted closely to the experimental values. The linear relation between the H+ coverage and enhancement of the reaction rate by the electric field are indicated clearly. These insights are fundamentally important for the further investigation of catalytic reactions in the electric field.
This study was supported by JST MIRAI.
Conflicts of interest
The authors have no conflict to declare related to this paper.
Notes and references
- A. Iwase and A. Kudo, Chem. Commun., 2017, 53, 6156–6159 RSC.
- J. Xu, C. Pan, T. Takata and K. Domen, Chem. Commun., 2015, 51, 7191–7194 RSC.
- F. Che, J. T. Gray, S. Ha and J. S. McEwen, ACS Catal., 2017, 7(10), 6957–6968 CrossRef CAS.
- A. Yamamoto, S. Mizuba, Y. Saeki and H. Yoshida, Appl. Catal., A, 2016, 521, 125–132 CrossRef CAS.
- M. Iwamoto, M. Akiyama, K. Aihara and T. Deguchi, ACS Catal., 2017, 7, 6924–6929 CrossRef CAS.
- Y. Kobayashi, N. Shimoda, Y. Kimura and Y. Satokawa, ECS Trans., 2017, 75(42), 43–52 CrossRef CAS.
- L. Zhang, L. X. Ding, G. F. Chen, X. Yang and H. Wang, Angew. Chem., Int. Ed., 2019, 58, 2612–2616 CrossRef CAS PubMed.
- T. Nozaki, N. Muta, S. Kado and K. Okazaki, Catal. Today, 2004, 89(1–2), 57–65 CrossRef CAS.
- M. Torimoto, K. Murakami and Y. Sekine, Bull. Chem. Soc. Jpn., 2019, 92(10), 1785–1792 CrossRef CAS.
- M. Torimoto, S. Ogo, D. Harjowinoto, T. Higo, J. G. Seo, S. Furukawa and Y. Sekine, Chem. Commun., 2019, 55, 6693–6695 RSC.
- S. Okada, R. Manabe, R. Inagaki, S. Ogo and Y. Sekine, Catal. Today, 2018, 307, 272–276 CrossRef CAS.
- R. Manabe, S. Okada, R. Inagaki, K. Oshima, S. Ogo and Y. Sekine, Sci. Rep., 2016, 6, 38007 CrossRef CAS PubMed.
- T. Yabe, K. Yamada, T. Oguri, T. Higo and Y. Sekine, ACS Catal., 2018, 8, 11470–11477 CrossRef CAS.
- R. Inagaki, R. Manabe, Y. Hisai, Y. Kamite, T. Yabe, S. Ogo and Y. Sekine, Int. J. Hydrogen Energy, 2018, 43(31), 14310–14318 CrossRef CAS.
- K. Takise, A. Sato, K. Muraguchi, S. Ogo and Y. Sekine, Appl. Catal., A, 2019, 573, 56–63 CrossRef CAS.
- M. Kosaka, T. Higo, S. Ogo, J. G. Seo, K. Imagawa, S. Kado and Y. Sekine, Int. J. Hydrogen Energy, 2020, 45(1), 738–743 CrossRef CAS.
- K. Takise, A. Sato, S. Ogo, J. G. Seo, K. Imagawa, S. Kado and Y. Sekine, RSC Adv., 2019, 9, 27743–27748 RSC.
- K. Takise, A. Sato, K. Murakami, S. Ogo, J. G. Seo, K. Imagawa, S. Kado and Y. Sekine, RSC Adv., 2019, 9, 5918–5924 RSC.
- K. Murakami, Y. Tanaka, S. Hayashi, R. Sakai, Y. Hisai, Y. Mizutani, A. Ishikawa, T. Higo, S. Ogo, J. G. Seo, H. Tsuneki, H. Nakai and Y. Sekine, J. Chem. Phys., 2019, 151, 064708 CrossRef.
- K. Murakami, Y. Tanaka, R. Sakai, K. Toko, K. Ito, A. Ishikawa, T. Higo, T. Yabe, S. Ogo, M. Ikeda, H. Tsuneki, H. Nakai and Y. Sekine, Catal. Today DOI:10.1016/j.cattod.2018.10.055.
- K. Murakami, R. Manabe, H. Nakatsubo, T. Yabe, S. Ogo and Y. Sekine, Catal. Today, 2018, 303, 271–275 CrossRef CAS.
- A. Gondo, R. Manabe, R. Sakai, K. Murakami, T. Yabe, S. Ogo, M. Ikeda, H. Tsuneki and Y. Sekine, Catal. Lett., 2018, 148(7), 1929–1938 CrossRef CAS.
- R. Manabe, H. Nakatsubo, A. Gondo, K. Murakami, S. Ogo, H. Tsuneki, M. Ikeda, A. Ishikawa, H. Nakai and Y. Sekine, Chem. Sci., 2017, 8, 5434–5439 RSC.
- K. Aika, A. Ohya, A. Ozaki, Y. Inoue and I. Yasumori, J. Catal., 1985, 92, 305–311 CrossRef CAS.
- H. Bielawa, O. Hinrichsen, A. Birkner and M. Muhler, Angew. Chem., Int. Ed., 2001, 40(6), 1061–1063 CrossRef CAS PubMed.
- S. E. Siporin and R. J. Davis, J. Catal., 2004, 225, 359–368 CrossRef CAS.
- Y. Niwa and K. Aika, Chem. Lett., 1996, 3–4 CrossRef CAS.
- K. Honkala, A. Hellman, I. N. Remediakis, A. Logadottir, A. Carlsson, S. Dahl, C. H. Christensen and J. K. Nørskov, Science, 2005, 307, 555–558 CrossRef CAS PubMed.
- K. Werner, X. Weng, F. Calaza, M. Sterrer, T. Kropp, J. Paier, J. Sauer, M. Wilde, K. Fukutani, S. Shaikhutdinov and H. J. Freund, J. Am. Chem. Soc., 2017, 139(48), 17608–17616 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0cc00482k |
|
| This journal is © The Royal Society of Chemistry 2020 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence a,
Shuhei
Ogo
a,
Shuhei
Ogo