
Nickel-catalyzed intramolecular desymmetrization addition of aryl halides to 1,3-diketones†
Pan
Zhou
and
Tao
XU
 *
*
Shanghai Key Laboratory of Chemical Assessment and Sustainability, School of Chemical Science and Engineering, Tongji University, 1239 Siping Road, Shanghai, 200092, P. R. China. E-mail: taoxu@tongji.edu.cn
First published on 12th March 2020
Abstract
A nickel-catalyzed intramolecular addition of aryl halides to 1,3-diketones was first developed. This desymmetrization reaction afforded polycyclic products bearing two tetrasubstituted centers with excellent diastereoselectivities and high yields. Moderate enantioselectivities were achieved in the presence of a chiral ligand. This transformation has great potential for the synthesis of polycyclic compounds including spiro[4,4,3,0] compounds.
Polycyclic frameworks with tetrasubstituted carbons, including tertiary alcohols and all-carbon quaternary centres, are widespread in natural products, organic materials, and synthetic biologically active molecules (Fig. 1).1 It has attracted many efforts towards exploring convenient and efficient protocols to access such frameworks, especially the construction of stereospecific quaternary centers.2 In particular, desymmetrization reactions have received much attention recently because of their numerous advantages for the construction of multi-chiral quaternary carbon centres as well as bicyclic skeletons with high stereoselectivities.3
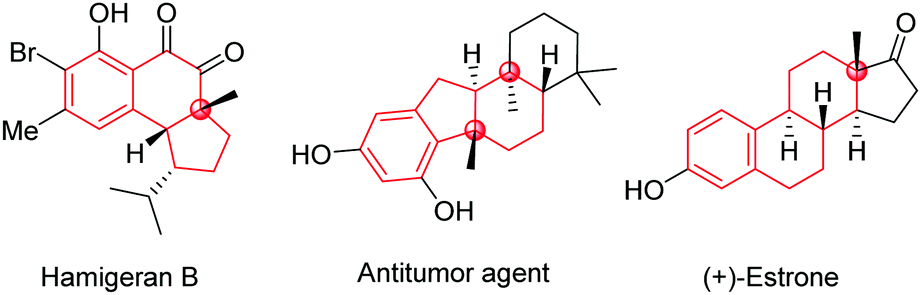 | ||
| Fig. 1 Selected examples with bicycle skeletons in natural products and drugs. | ||
Cyclic symmetric 1,3-diketones with two different substituents at the prochiral carbon are one of the most useful scaffolds to demonstrate the power of desymmetrization, such as the Hajos-Parrish reaction.4 Transition-metal catalysts are often used to improve the efficiencies and selectivities of desymmetrization reactions.5 Although many transformations involving C(sp3)–metal intermediates with a 1,3-diketone partner have been reported, reactions with C(sp2)–metal species have not been studied comprehensively.6,7 In 2017, Shi's group published an enantioselective palladium-catalyzed ketone α-arylative desymmetrization of 1,3-diketones (eqn (1), Scheme 1).8 Besides, Lam and co-workers reported a nickel-catalyzed addition reaction of a vinyl-nickel intermediate, which was generated in situ from an arylboronic acid with an alkyne, to the carbonyl site (eqn (2), Scheme 1).9 Although enantioselective desymmetrization Heck or Michael reactions initiated from aryl or vinyl halides have been developed,10 the desymmetrization addition of aryl halides to diketones has not been disclosed.
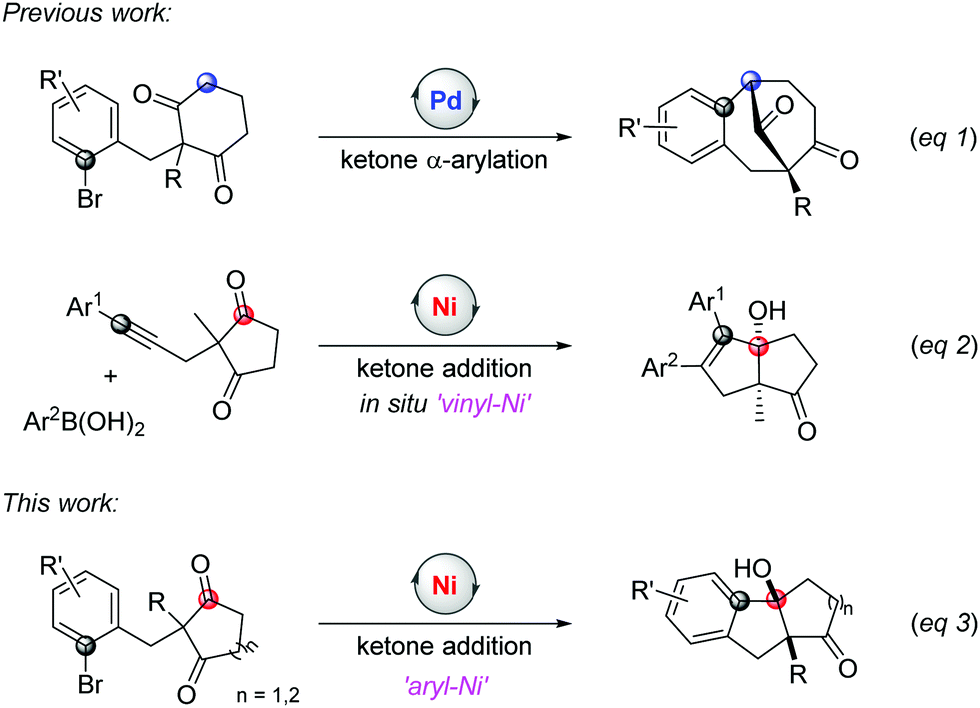 | ||
| Scheme 1 The desymmetrization reactions with vinyl-metal intermediates. | ||
The addition pathway of an aryl framework to ketones is a convenient route to access tertiary benzylic alcohols and usually relies on organometallic reagents, such as Grignard or organozinc reagents.11 These methods are widely used because of the ready availability of aryl halide materials but also face some issues when sensitive functional groups are involved. Therefore, efforts are taken to develop a superior alternative where aryl halides are directly employed in the presence of a transition metal catalyst and reductant.12,13 This approach can efficiently avoid the use of dangerous organometallic reagents with low functional group tolerance. For example, the palladium-catalyzed addition of aryl bromides to ketones was reported by Yamamoto et al. in 2000.14 Recently, Kündig's and Jia's groups reported the reaction of aryl halides or vinyl bromides with α-ketoamides.15 Considering the potential of merging desymmetrization and ketone addition strategies to extend chiral tetrasubstituted carbons, herein, we report a nickel-catalyzed addition of aryl halides to 1,3-diketones to access the bicyclic[m.n.0] skeleton with high yields and excellent diastereoselectivities (eqn (3), Scheme 1). In addition, the enantioselective desymmetrization of this transformation is investigated to give two stereospecific tetrasubstituted carbon centres with moderate enantioselectivities in the presence of a chiral ligand.
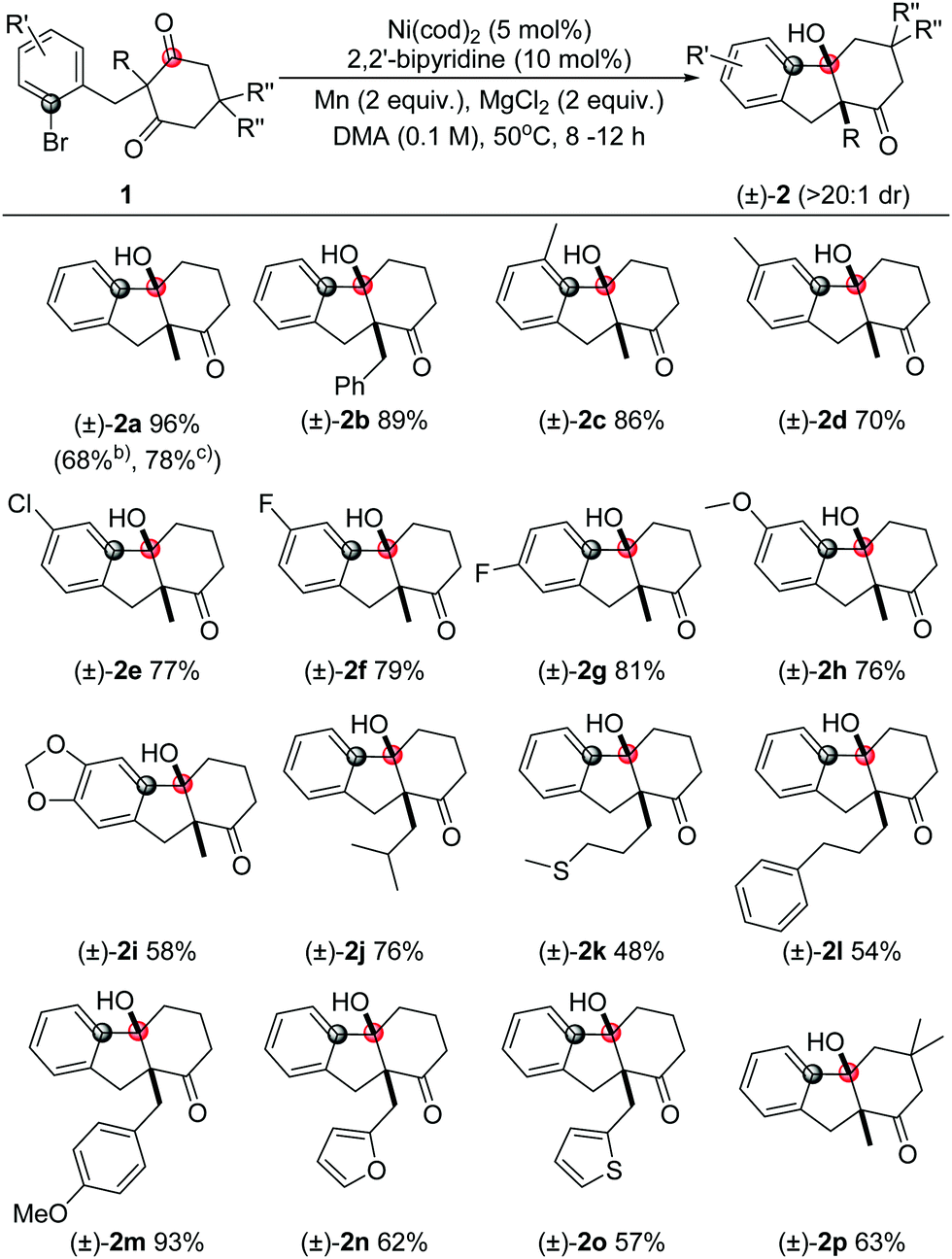
We commenced this study with 2-(2-bromobenzyl)-2-methylcyclohexane-1,3-dione (1a) as a model substrate, under the conditions with nickel as a catalyst, and metal dust as the reductant (Table 1). After careful evaluations of the ligands, reductants, and solvents (for details, see the ESI†), we were delighted to find that product 2a can be afforded in very high yield and excellent diastereoselectivity (dr >20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) under these conditions as illustrated in entry 1, Table 1. The yield decreased a little bit when conducting at room temperature (entry 2) or lower catalyst loading (entry 3). Other ligands, such as 1,10-phenanthroline, 1,2-bis(diphenylphosphino)ethane, were also suitable but with lower yields (entries 4 and 5). Other catalysts, such as FeBr3, CoBr2, and Cu(OTf)2, did not afford the desired products, while palladium acetate only gave a poor conversion (entries 6–9). Additionally, control experiments showed that the nickel catalyst, ligand, and reductant were all crucial to the ketone addition reaction (entries 10–12).
:1) under these conditions as illustrated in entry 1, Table 1. The yield decreased a little bit when conducting at room temperature (entry 2) or lower catalyst loading (entry 3). Other ligands, such as 1,10-phenanthroline, 1,2-bis(diphenylphosphino)ethane, were also suitable but with lower yields (entries 4 and 5). Other catalysts, such as FeBr3, CoBr2, and Cu(OTf)2, did not afford the desired products, while palladium acetate only gave a poor conversion (entries 6–9). Additionally, control experiments showed that the nickel catalyst, ligand, and reductant were all crucial to the ketone addition reaction (entries 10–12).
|
|
|||
|---|---|---|---|
| Entry | Substrate | Catalyst | Yield |
|
a General conditions: 1 (0.1 mmol), catalyst (5 mol%), 2,2′-bipyridine (10 mol%), Mn (0.2 mmol), DMA (1.0 mL), 50 °C, 8–12 h; yields and ratios (dr >20:1) were determined by NMR by using CH2Br2 as the internal standard.
b At room temperature.
c Ni(cod)2 (2 mol%), 1,1-bipyridine (5 mol%).
d 1,10-Phenanthroline (10 mol%) as the ligand.
e 1,2-Bis(diphenylphosphino)ethane (10 mol%) as the ligand.
f Without the ligand.
g Without Mn.
h NaI (0.2 mmol) was added as the additive.
i MgCl2 (0.2 mmol) was added as the additive.
|
|||
| 1 | 1a | Ni(cod)2 | (±)-2a(>99%) |
| 2b | 1a | Ni(cod)2 | (±)-2a(95%) |
| 3c | 1a | Ni(cod)2 | (±)-2a(98%) |
| 4d | 1a | Ni(cod)2 | (±)-2a(72%) |
| 5e | 1a | Ni(cod)2 | (±)-2a(70%) |
| 6 | 1a | FeBr3 | n.r. |
| 7 | 1a | CoBr2 | n.r. |
| 8 | 1a | Cu(OTf)2 | n.r. |
| 9 | 1a | Pd(OAc)2 | (±)-2a(27%) |
| 10 | 1a | — | n.r. |
| 11f | 1a | Ni(cod)2 | n.r. |
| 12g | 1a | Ni(cod)2 | n.r. |
| 13 | 1b | Ni(cod)2 | (±)-2b(46%) |
| 14h | 1b | Ni(cod)2 | (±)-2b(84%) |
| 15i | 1b | Ni(cod)2 | (±)-2b(>99%) |
| 16 | 1b | — | n.r. |

However, when the benzyl substituted substrate 1b was employed, only a moderate yield was detected (entry 13, Table 1). To improve the yield, various additives were studied. The reaction gave 84% and 99% yields in the presence of 2 equivalent of NaI or MgCl2, respectively (entries 14 and 15). MgCl2 was often employed to improve the yields in reductive coupling reactions and believed to facilitate the reduction of the metal complex by Zn or Mn to generate active metal catalysis.16 Meanwhile, the Lewis acidity of MgCl2 may further activate the carbonyl group to favour the addition step,17 as ZnBr2 in the aldehyde addition demonstrated by the Weix group.13b However, the exact role of MgCl2 still remains elusive. The nickel catalyst was also indispensable under these conditions as well (entry 16).
With the optimal conditions in hand (entry 15, Table 1), we next explored the scope of this transformation (representative examples are summarized in Table 2). In general, the substrates with either electron-neutral, -donating or -withdrawing substituents, as well as groups on various positions of the aryl part, can proceed smoothly to access the corresponding products in high yields and excellent diastereoselectivities (>20:1 dr). The syn configuration was confirmed by X-ray crystallography using compound 2a (Fig. 2).18 The reaction on 1 mmol scale gave a moderate yield. The aryl iodide substrate can also be used but aryl chloride did not form any target product. The alkyl substituents on the aryl group did not significantly affect the reactivity (2c, 2d), even in the adjacent site (2c). The aryl fluoride or chloride groups (2e–2g), and the methoxide (2k, 2i) can survive under these nickel-catalyzed reactions. More investigations were focused on the quaternary carbon centre of the 1,3-diketone part. A variety of functional groups such as alkyl (2j, 2l), sulfur ether (2k), and benzyl (2b, 2m) were well tolerated. Importantly, heterocyclic structures consisting of furan (2n) and thiophene (2o) were also suitable substrates. The substituents on the 1,3-diketones framework have no obvious effect on the yield (2p).
|
a General conditions: 1 (0.2 mmol), Ni(cod)2 (5 mol%), 2,2′-bipyridine (10 mol%), Mn (0.4 mmol), MgCl2 (0.4 mmol), DMA (2.0 mL), 50 °C, 8–12 h; isolated yields were given. For all tested samples, the products with >20:1 dr were found by 1H NMR of the crude mixture.
b On 1 mmol scale.
c Aryl iodide substrate was used.
|
|---|
|
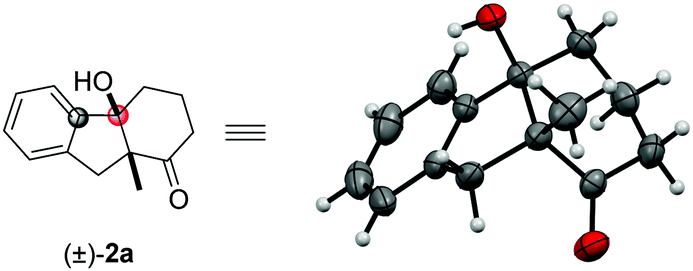 | ||
| Fig. 2 The X-ray crystallography of compound 2a. | ||
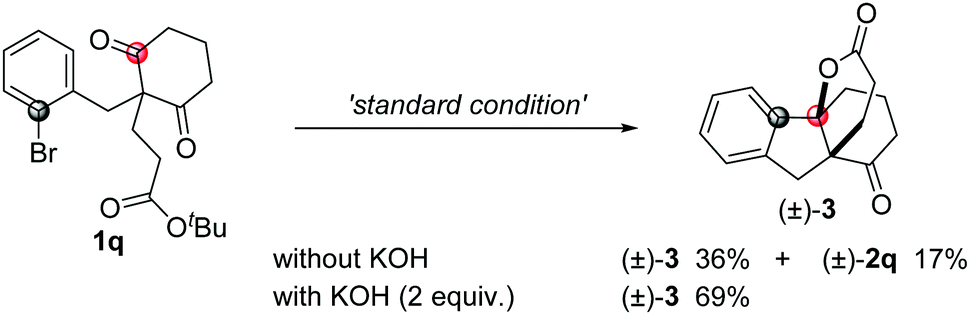
To our surprise, when compound 1q was used in the reaction, besides the normal product 2q (17%), another spiro[4,4,3,0] compound 3 was obtained in 36% yield with high diastereoselectivity, which was possibly produced through the desymmetrization addition followed by a formal intramolecular transesterification process. The yield of this spiro compound increased to 69% yield in the presence of KOH as an additive (eqn (4)).19
To further study the scopes, some other types of substrates were studied. It was found that as well as the cyclohexane-1,3-diketones, substituted cyclopentane-1,3-diketone substrates also afforded the desired products under the conditions (5a, eqn (5)). In addition, the scope of this desymmetrization reaction could be extended to prepare products with [4,4,0] skeletons in high yield by increasing the carbon linker number (5b, eqn (6)). Interestingly, an acyclic substrate was also effective to construct a multi-substituted 2,3-dihydro-1H-indene although the diastereoselectivity was not as high as that obtained for the cyclic substrates (5c, eqn (7)).
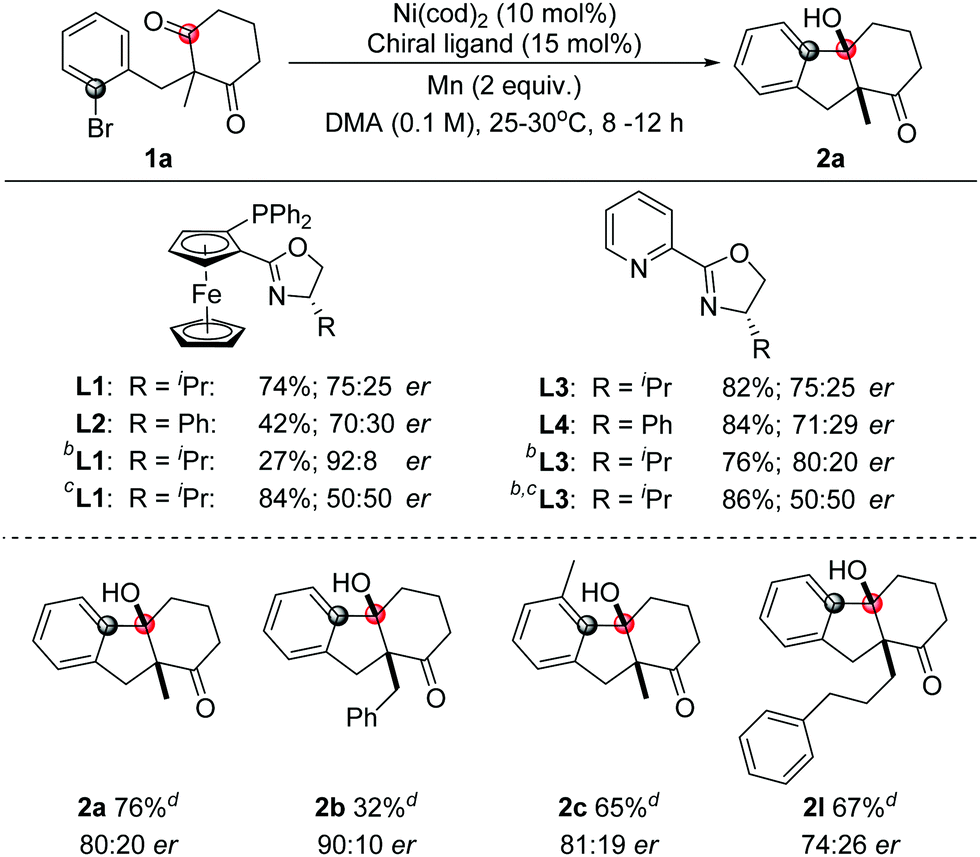
To investigate the enantioselective transformation of this reaction, some preliminary efforts were taken on the asymmetric reaction. Considering that oxazoline ligands were widely used in the nickel-catalyzed reactions, this type of ligand was studied. The use of a chiral ligand L1 or L3 could give 75:25 er. Similar results were obtained in the presence of L2 or L4. The product was afforded in 92:8 er with L1 as a ligand in the mixed DMA/1,4-dioxane solvent but the yield was very low. To improve the yield, MgCl2 was added. However, the reaction showed no enantioselectivity then. The use of ligand L3 in the mixed solvents could give a better yield and moderate enantioselectivity synchronously. Under these conditions, moderate yields and enantioselectivities were obtained with some tested samples as shown in Table 3.
 | (4) |
 | (5) |
 | (6) |
 | (7) |
|
a General conditions: 1a (0.1 mmol), Ni(cod)2 (10 mol%), chiral ligand (15 mol%), Mn (0.2 mmol), DMA (1.0 mL), 25–30 °C, 8–12 h; yields and ratios (dr >20:1) were determined by NMR by using CH2Br2 as the internal standard, and the ee values were determined by chiral HPLC analysis.
b DMA/1,4-dioxone (v/v 4/1) was used as the solvent.
c MgCl2 (0.2 mmol) was added as the additive.
d Isolated yield was given and the absolute stereochemistry was not determined.
|
|---|
|
Bicycle product 2a obtained by this method was used to demonstrate the synthetic utility of this methodology (Scheme 2). The carbonyl group can be reduced by NaBH4 to give the alcohol product (6a) with high diastereoselectivity. The epoxy unit was installed by Me3SI with high dr and good yield. The Wittig reaction can be employed to synthesize the olefin product (6c). Also, the tertiary alcohol can be transformed into the internal alkene product (6d) in the presence of p-TsOH.
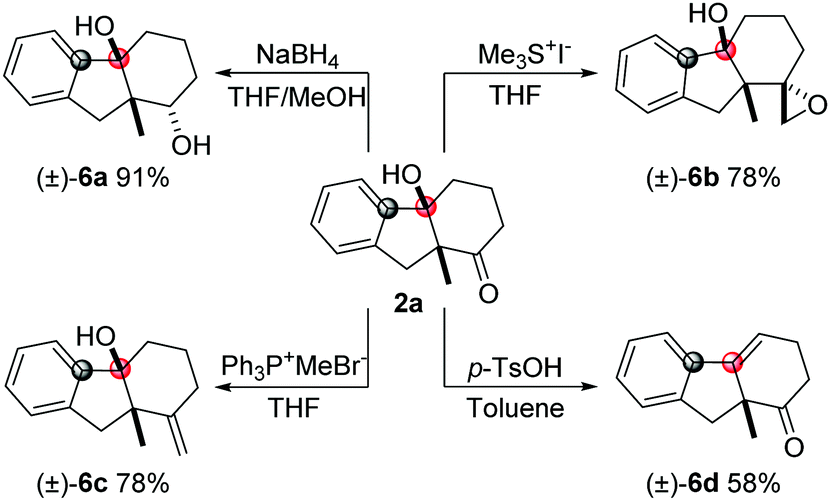 | ||
| Scheme 2 Derivatization of bicyclic product 2a. | ||
In summary, we have reported a nickel-catalyzed intramolecular addition of aryl halides to ketones, which produced a polycyclic framework with two tetrasubstituted carbons in excellent diastereoselectivity. Further attempts of the asymmetric reaction achieved moderate enantioselectivities. This novel desymmetrization reaction exhibits excellent functional group tolerance and high potential synthetic utility. Using this method, a spiro[4,4,3,0] compound could be easily obtained in good yield.
We thank the NFS of China (Grants 21801188), the Shanghai Pujiang Program, the “1000 Youth Talents Plan”, the Fundamental Research Funds for the Central Universities, China and Tongji University for financial support. We thank Dr Natasha Lundin for polishing the manuscript.
Conflicts of interest
The authors declare no competing financial interest.Notes and references
- (a) K. C. Nicolaou, D. Gray and J. Tae, Angew. Chem., Int. Ed., 2001, 40, 3675–3678 CrossRef CAS; (b) B. M. Trost, C. Pissot-Soldermann, I. Chen and G. M. Schroeder, J. Am. Chem. Soc., 2004, 126, 4480–4481 CrossRef CAS PubMed; (c) L. G. Meimetis, M. Nodwell, L. Yang, X. Wang, J. Wu, C. Harwig, G. R. Stenton, L. F. Mackenzie, T. MacRury, B. O. Patrick, A. Ming-Lum, C. J. Ong, G. Krystal, A. L.-F. Mui and R. J. Andersen, Eur. J. Org. Chem., 2012, 5195–5207 CrossRef CAS; (d) R. Andersen, M. Nodwell and A. Mui, WO2007147252, 2007.
- For some selected reviews, see: (a) A. Y. Hong and B. M. Stoltz, Eur. J. Org. Chem., 2013, 2745–2759 CrossRef CAS PubMed; (b) K. W. Quasdorf and L. E. Overman, Nature, 2014, 516, 181–191 CrossRef CAS PubMed; (c) P. G. Cozzi, R. Hilgraf and N. Zimmermann, Eur. J. Org. Chem., 2007, 5969–5994 CrossRef CAS.
- (a) X.-P. Zeng, Z.-Y. Cao, Y.-H. Wang, F. Zhou and J. Zhou, Chem. Rev., 2016, 116, 7330–7396 CrossRef CAS PubMed; (b) K. S. Petersen, Tetrahedron Lett., 2015, 56, 6523–6535 CrossRef CAS PubMed.
- (a) Z. G. Hajos and D. R. Parrish, DE2102623, 1971; (b) U. Eder, G. Sauer and R. Wiechert, Angew. Chem., Int. Ed. Engl., 1971, 10, 496–497 CrossRef CAS; (c) S. Prévost, N. Dupré, M. Leutzsch, Q. Wang, V. Wakchaure and B. List, Angew. Chem., Int. Ed., 2014, 53, 8770–8773 CrossRef PubMed.
- (a) Q. Gong, J. Wen and X. Zhang, Chem. Sci., 2019, 10, 6350 RSC; (b) B. Shi, S. Merten, D. K. Y. Wong, J. C. K. Chu, L. L. Liu, S. K. Lam, A. Jäger, W.-T. Wong, P. Chiu and P. Metz, Adv. Synth. Catal., 2009, 351, 3128–3132 CrossRef CAS; (c) B. M. Partridge, J. Solana González and H. W. Lam, Angew. Chem., Int. Ed., 2014, 53, 6523–6527 CrossRef CAS PubMed.
- (a) B. M. Bocknack, L.-C. Wang and M. J. Krische, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 5421–5424 CrossRef CAS PubMed; (b) A. R. Burns, J. Solana González and H. W. Lam, Angew. Chem., Int. Ed., 2012, 51, 10827–10831 CrossRef CAS PubMed; (c) J. Deschamp and O. Riant, Org. Lett., 2009, 11, 1217–1220 CrossRef CAS PubMed; (d) J. Ou, W.-T. Wong and P. Chiu, Org. Biomol. Chem., 2012, 10, 5971–5978 RSC.
- P. Zheng, X. Han, J. Hu, X. Zhao and T. Xu, Org. Lett., 2019, 21, 6040–6044 CrossRef CAS PubMed.
- C. Zhu, D. Wang, Y. Zhao, W.-Y. Sun and Z. Shi, J. Am. Chem. Soc., 2017, 139, 16486–16489 CrossRef CAS PubMed.
- C. Clarke, C. A. Incerti-Pradillos and H. W. Lam, J. Am. Chem. Soc., 2016, 138, 8068–8071 CrossRef CAS PubMed.
- For some selected samples, see: (a) T. Ohshima, K. Kagechika, M. Adachi, M. Sodeoka and M. Shibasaki, J. Am. Chem. Soc., 1996, 118, 7108–7116 CrossRef CAS; (b) Y. Sato, M. Sodeoka and M. Shibasaki, J. Org. Chem., 1989, 54, 4738–4739 CrossRef CAS; (c) P. Liu, Y. Fukui, P. Tian, Z.-T. He, C.-Y. Sun, N.-Y. Wu and G.-Q. Lin, J. Am. Chem. Soc., 2013, 135, 11700–11703 CrossRef CAS PubMed.
- For some selected reviews, see: (a) Y.-L. Liu and X.-T. Lin, Adv. Synth. Catal., 2019, 361, 876–918 CrossRef CAS; (b) J. F. Collados, R. Solà, S. R. Harutyunyan and B. Maciá, ACS Catal., 2016, 6, 1952–1970 CrossRef CAS; (c) J. Rong, T. Pellegrini and S. R. Harutyunyan, Chem. – Eur. J., 2016, 22, 3558–3570 CrossRef CAS PubMed.
- For some selected samples, see: (a) R. Shi and X. Hu, Angew. Chem., Int. Ed., 2019, 58, 7454–7458 CrossRef CAS PubMed; (b) H. Chen, X. Jia, Y. Yu, Q. Qian and H. Gong, Angew. Chem., Int. Ed., 2017, 56, 13103–13106 CrossRef CAS PubMed; (c) K. E. Poremba, N. T. Kadunce, N. Suzuki, A. H. Cherney and S. E. Reisman, J. Am. Chem. Soc., 2017, 139, 5684–5687 CrossRef CAS PubMed.
- For some reactions on aldehyde addition, see: (a) K. K. Majumdar and C.-H. Cheng, Org. Lett., 2000, 2, 2295–2298 CrossRef CAS PubMed; (b) K. J. Garcia, M. M. Gilbert and D. J. Weix, J. Am. Chem. Soc., 2019, 141, 1823–1827 CrossRef CAS PubMed; (c) S. Ishida, H. Suzuki, S. Uchida, E. Yamaguchi and A. Itoh, Eur. J. Org. Chem., 2019, 7483–7487 CrossRef CAS.
- (a) L. G. Quan, M. Lamrani and Y. Yamamoto, J. Am. Chem. Soc., 2000, 122, 4827–4828 CrossRef CAS; (b) D. Solé, L. Vallverdú, E. Peidró and J. Bonjoch, Chem. Commun., 2001, 1888–1889 RSC.
- (a) J.-Q. He, C. Chen, W.-B. Yu, R.-R. Liu, M. Xu, Y.-J. Li, J.-R. Gao and Y.-X. Jia, Tetrahedron Lett., 2014, 55, 2805–2808 CrossRef CAS; (b) J.-X. Hu, H. Wu, C.-Y. Li, W.-J. Sheng, Y.-X. Jia and J.-R. Gao, Chem. – Eur. J., 2011, 17, 5234–5237 CrossRef CAS; (c) Y.-X. Jia, D. Katayev and E. P. Kündig, Chem. Commun., 2010, 46, 130–132 RSC.
- For some selected samples, see: (a) Y. Ye, H. Chen, J. L. Sessler and H. Gong, J. Am. Chem. Soc., 2019, 141, 820–824 CrossRef CAS PubMed; (b) H. Chen, X. Jia, Y. Yu, Q. Qian and H. Gong, Angew. Chem., Int. Ed., 2017, 56, 13103–13106 CrossRef CAS PubMed; (c) W. Yu, L. Chen, J. Tao, T. Wang and J. Fu, Chem. Commun., 2019, 55, 5918–5921 RSC; (d) X. Wang, G. Ma, Y. Peng, C. E. Pitsch, B. J. Moll, T. D. Ly, X. Wang and H. Gong, J. Am. Chem. Soc., 2018, 140, 14490–14497 CrossRef CAS PubMed.
- (a) R. W. Coscia and T. H. Lambert, J. Am. Chem. Soc., 2009, 131, 2496–2498 CrossRef CAS PubMed; (b) X. Fang, K. Liu and C. Li, J. Am. Chem. Soc., 2010, 132, 2271–2283 Search PubMed; (c) D. Yang, Q. Gao, C.-S. Lee and K.-K. Cheung, Org. Lett., 2002, 4, 3271–3274 CrossRef CAS PubMed.
- CCDC 1951717 contains the supplementary crystallographic data for this paper [2a]†.
- KOH could be a promoter for the intramolecular transesterification. For some literatures, see: (a) P.-Q. Huang and H. Geng, Chem. Sci., 2018, 20, 593–599 CAS; (b) M. J. Batchelor, R. J. Gillespie, J. M. C. Golec, C. J. R. Hedgecock, S. D. Jones and R. Murdoch, Tetrahedron, 1994, 50, 809–826 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1951717. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0cc00457j |
| This journal is © The Royal Society of Chemistry 2020 |