
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Consecutive 5′- and 3′-amide linkages stabilise antisense oligonucleotides and elicit an efficient RNase H response†
Sven
Epple
 a,
Cameron
Thorpe
a,
Ysobel R.
Baker
a,
Afaf H.
El-Sagheer
ab and
Tom
Brown
*a
a,
Cameron
Thorpe
a,
Ysobel R.
Baker
a,
Afaf H.
El-Sagheer
ab and
Tom
Brown
*a
aChemistry Research Laboratory, University of Oxford, Oxford, OX1 3TA, UK. E-mail: tom.brown@chem.ox.ac.uk
bChemistry Branch, Department of Science and Mathematics, Faculty of Petroleum and Mining Engineering, Suez University, Suez 43721, Egypt
First published on 15th April 2020
Abstract
Antisense oligonucleotides are now entering the clinic for hard-to-treat diseases. New chemical modifications are urgently required to enhance their drug-like properties. We combine amide coupling with standard oligonucleotide synthesis to assemble backbone chimera gapmers that trigger an efficient RNase H response while improving serum life time and cellular uptake.
Antisense oligonucleotides (ASOs) are short (∼20mer) chemically modified oligomers that bind to their complementary RNA targets to modulate gene expression at the mRNA level.1,2 Thus, ASOs can target proteins that are considered undruggable through conventional approaches.3 As such, they hold enormous promise for hard-to-treat diseases as evidenced by a number of recently approved oligonucleotide (ON)-based drugs.4–6
Chemical modifications are essential to improve the serum stability and pharmacodynamic properties of ASOs, as unmodified ONs are rapidly digested by nucleases in vivo7,8 and suffer from poor cellular uptake and tissue distribution.9 Whilst there have been considerable advances to modify ONs either at the nucleobase, sugar or backbone, a set of distinct chemical modifications to confer ideal drug-like properties has not yet been achieved.10 Commonly used ribose modifications include 2′-F, 2′-OMe, 2′-O-(2-methoxyethyl) and locked nucleic acids, all of which have been shown to improve target affinity and serum stability.11 The most commonly used phosphodiester (PO) mimic is the phosphorothioate (PS) linkage which is compatible with ribonuclease H (RNase H) activation,12 a mechanism resulting in degradation of an mRNA upon formation of an ASO:mRNA heteroduplex. PS linkages also improve metabolic stability13 and enhance pharmacodynamic properties through interactions with plasma proteins.8,14,15 However, unspecific protein binding can contribute to the toxic potential of PS-ASOs16,17 and the PS linkage is P-chiral resulting in a mixture of diastereomers (more than half a million isomers in Mipomersen12,18). Moreover, inefficient cellular uptake remains a major challenge for ASO therapeutics. Therefore, the investigation of other artificial backbone linkages is urgently needed.
Charge-neutral backbone modifications represent an interesting class of PO mimics. Among those, (thio)phosphonoacetate esters,19 phosphotriesters20 and alkyl phosphonates21 can enhance cellular uptake by eliminating the PO negative charges. Moreover, incorporation of a single methylphosphonate can eliminate hepatotoxicity of PS-ASOs.22 Phosphorodiamidate morpholino oligomers (PMOs) combine backbone and sugar modifications and enhance delivery through interactions with scavenger receptors,23 but are not compatible with standard ON synthesis. All aforementioned PO mimics also suffer from increased steric complexity due to their P-chiral linkages.
The absence of a chiral centre and the well-established solid-phase peptide synthesis methods make the amide internucleoside linkage24 (AM, Fig. 1A) a promising candidate for backbone surrogates. Within DNA, isolated amides can slightly increase duplex stability with target RNA24 while consecutive amides were reported to have minimal effects.25 Amide-modified ONs are stable in serum24 and the backbone is well tolerated in replication, transcription26 and RNAi27 but its application in the context of RNase H activation has not been reported.
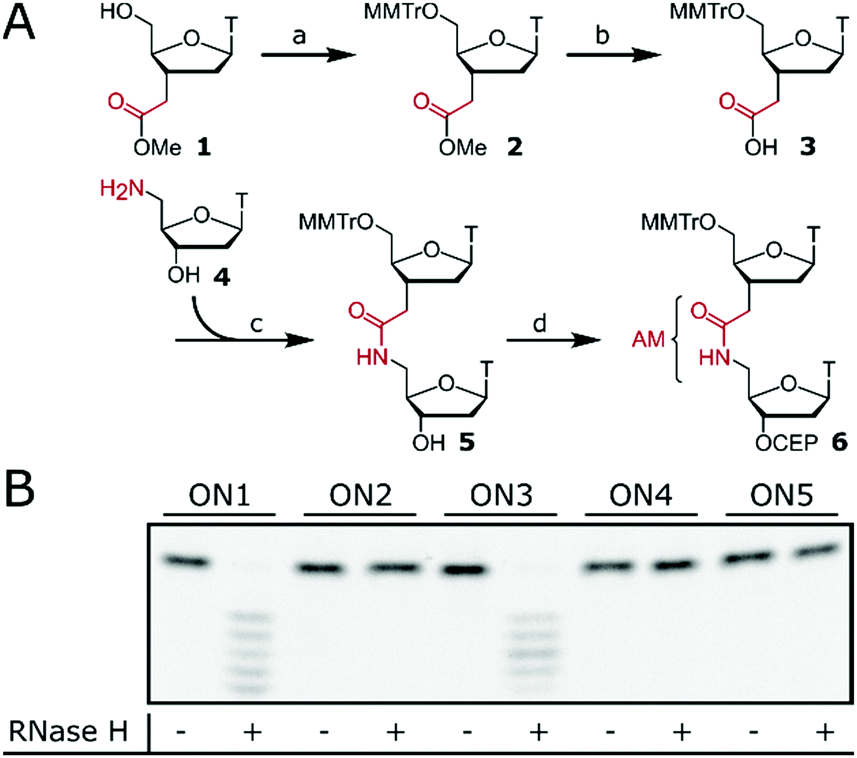 | ||
Fig. 1 (A) Synthesis of phosphoramidite 6 as a building block in ON synthesis. Reagents and conditions (a) MMTr-Cl, pyridine (py), rt, 82%; (b) LiOH, THF/H2O (3/1), 60 °C, 81%; (c) 1-[bis(dimethylamino)methylene]-1H-1,2,3-triazolo[4,5-b]pyridinium 3-oxid hexa-fluorophosphate (HATU), N,N-diisopropylethylamine (DIPEA), DMF, rt, 69%; (d) chloro-(diiso-propylamino)-β-cyanoethoxyphosphine, DIPEA, CH2Cl2, rt, 56%. CEP = cyanoethyl-phosphoramidite, MMTr = 4-methoxytriphenylmethyl, T = thymine. (B) Degradation of FL-RNA1 by Escherichia coli RNase H after incubation with ASOs at rt for 20 h. ASOs are ON1–ON5. ASO/FL-RNA1, 1.1/1 (mole/mole). ON1: 5′-T12-3′; ON2: 5′-TpTpTaTpTpTaTpTp TaTpTpT-3′; ON3: 5′-![[U with combining low line]](https://www.rsc.org/images/entities/char_0055_0332.gif) pppTpTpTpTpTpTppp-3′; ON4: 5′-pppTpTpTaTpTpTppp-3′; ON5: 5′-pppTpTaTpTpTaTppp-3′; FL-RNA1: 5′-FL-A12-3′. a = AM, FL = fluorescein, p = PO, T = thymidine, = 2′-OMe uridine. pppTpTpTpTpTpTppp-3′; ON4: 5′-pppTpTpTaTpTpTppp-3′; ON5: 5′-pppTpTaTpTpTaTppp-3′; FL-RNA1: 5′-FL-A12-3′. a = AM, FL = fluorescein, p = PO, T = thymidine, = 2′-OMe uridine. | ||
Here we report the introduction of isolated and consecutive amide linkages into ONs for RNase H-based antisense applications. We discuss possible ASO designs that contain the AM backbone to induce RNA target degradation and we evaluate serum stability and cellular uptake of a partially uncharged AM-gapmer ON.
Isolated AM internucleoside linkages were introduced via dinucleotide phosphoramidite 6 (Fig. 1A) with an internal amide as the building block for standard ON synthesis. Preparation of phosphoramidite 6 required 5′-tritylation of monomer 125 followed by ester hydrolysis to obtain acid 3. Amide coupling with amine 428 resulted in dimer 5 and subsequent phosphitylation gave 6 which can be used in standard ON synthesis.24 Phosphoramidite 6 was then used to synthesise ASOs (ON2, ON4, ON5, Fig. 1B) containing isolated AM linkages at various positions. The design of the tested ASOs was based on three consecutive interactions inside the PO binding pocket of RNase H with the ASO.12,29 ON2 contains three isolated AM linkages which are interspaced by two consecutive POs. Efficient RNase H response mediated by ON2 would confirm that the AM linkage could be accommodated inside the binding pocket of RNase H. The same rational was applied to ON4 and ON5 whose gapmer design further narrows the window for RNase H activation. ON1 (dT12) and gapmer ON3 with flanking 2′-OMe wings and a central dT6 region served as positive controls. ON1–ON5 were then tested to induce RNase H-mediated degradation of a fluorescein (FL)-labelled target RNA (FL-RNA1). However, no cleavage of the target RNA was observed for ASOs containing the AM linkage (ON2, ON4 and ON5), suggesting that the AM linkage is not tolerated within the PO binding pocket of RNase H.
AM–PO chimeras with sections of consecutive AM linkages were synthesised by adapting published protocols (Fig. 2).25,30–32 AM-coupling (i) of acid 725 to a 5′-amine forms the AM bond and deprotection (ii) and successive AM-coupling (i) of monomer 7 builds up sections of nucleosides consecutively linked through the AM linkage (AM-cycle). Introduction of a 5′-OH was achieved by AM-coupling (i) of monomer 3 and subsequent deprotection (ii). ON synthesis using standard phosphoramidite monomers builds up sections with PO linkages (PO-cycle). Transition from the PO-cycle to the AM-cycle can be achieved by PO-coupling (iii) of commercially available phosphoramidite 8 followed by oxidation (iv) and detritylation (ii) to introduce a 5′-amine as a substrate for the AM-cycle as described before. Using PyBOP and NMM as the coupling agent and base gave minimal side products and the combined PO- and AM-cycles gave chimeric gapmer ON6 with charge-neutral wings in an overall isolated yield of 16% (Fig. 3 and Fig. S1, S2, ESI†). The crude AM-modified ONs gave clean chromatographic traces (Fig. 3A and D) with the main peak corresponding to the desired product which was confirmed following purification (Fig. 3B and E) and mass analysis (Fig. 3C and F). We rationalise that the absence of a 2′-fuctionality can increase coupling efficiencies compared to consecutive amide couplings of more challenging RNA-type monomers.31,32
 | ||
| Fig. 2 Transition between PO- and amide-based ON synthesis. (i) AM-coupling: 4-methyl-morpholine (NMM, 32 equiv.), (benzotriazol-1-yloxy)tripyrrolidinophosphoniumhexa-fluorophosphate (PyBOP) (10 equiv.), acid 7 or 3 (10 equiv.), DMF, rt, 2 h; (ii) de-protection: 3% (w/v) trichloroacetic acid in CH2Cl2, rt; (iii) standard PO-coupling: standard phosphoramidites or phosphoramidite 8, 5-(ethylthio)-1H-tetrazole, MeCN, rt; (iv) oxidation: 0.1 M I2, THF, py, H2O, rt. | ||
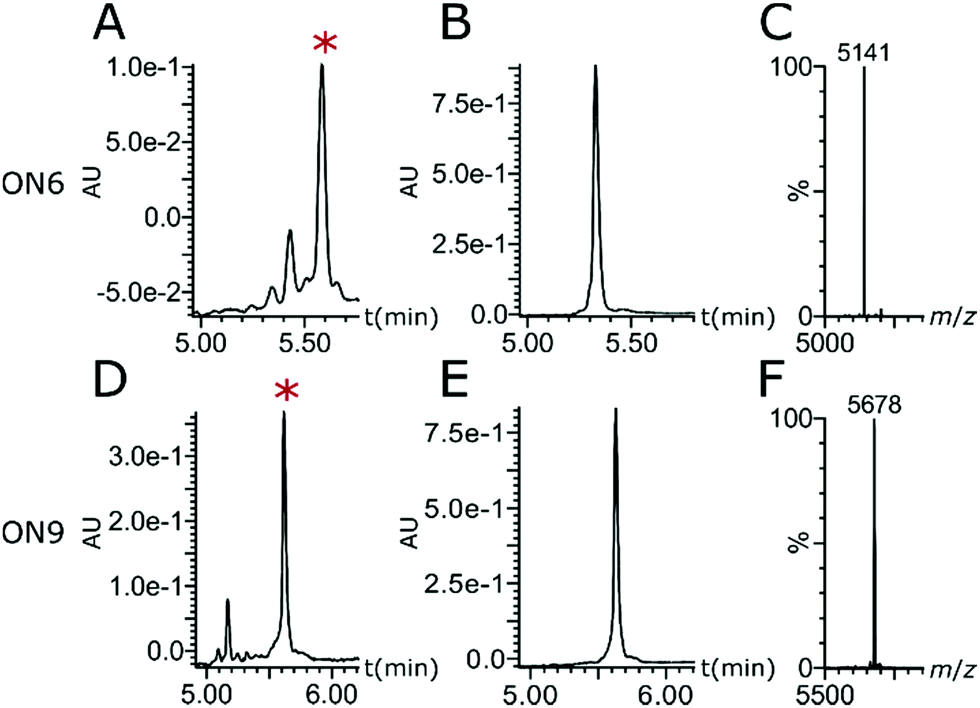 | ||
| Fig. 3 (A and D) Reversed-phase ultra performance liquid chromatography (RP-UPLC) traces of crude ONs. (B and E) RP-UPLC traces of purified ONs. (C and F) Mass analysis. ON6: 5′-TaTaTaTaTpCpCpTpGpApTpApGpTaTaTaTaT-3′; ON9: 5′-FLpTaTaTaTaTpCpCpTpGpApTpApGpTaTaTaTaT-3′. *Peak corresponding to desired ON. For full traces see Fig. S1 and S2 (ESI†). | ||
The consecutive AM backbones in ON6 constitute the flanking wings of the ASO, a commonly used strategy to retain RNase H activity while utilising the properties of otherwise RNase H-incompatible modifications, including the 2′-OMe modification33 (ON3, Fig. 1B). However, this strategy has not yet been reported for AM backbones. Thus, ON6 was tested in the RNase H assay while ON7 and ON8 (Fig. 4 and Table 1) were used as positive controls to induce degradation of a 5′-FL-labelled complementary target RNA (FL-RNA2). Aliquots of the reaction buffers containing E. coli RNase H, FL-RNA2 and a catalytic amount of ASOs (ON6, ON7 or ON8) were quenched at different time points and analysed by gel electrophoresis (Fig. 4 and Fig. S3, ESI†). The gels show that all tested ASOs induce complete target degradation within 30 min at 37 °C. Quantification confirmed that AM-gapmer ON6 activates RNase H as efficiently as the controls ON7 and ON8. This is a clear improvement on previously reported gapmers with charge-neutral wings using peptide nucleic acids (PNAs) which only induce target degradation in a non-catalytic way.34 Watts et al. reported that improved gapmer designs and optimisation of linkers between PNA and DNA sections can lead to catalytic activity but the charge-neutral section was limited to only one wing.35 In comparison, AM-gapmer ON6 shows efficient catalytic degradation of the target RNA.
 | ||
| Fig. 4 Degradation of FL-RNA2 by RNase H after incubation with ASOs at 37 °C over time. ASOs are ON6–ON8. ASO/FL-RNA2, 1/4 (mole/mole). Time points are 0 min, 1 min, 5 min, 15 min, 30 min and 1 h. Quantification of full-length FL-RNA2 was performed from gels using ImageJ. n = 3 ± standard error of the mean. FL-RNA2: 5′-FL-AAAAACUAUCAGGAAAAA-3′. Full gel images are shown in Fig. S3 (ESI†). For sequences see Table 1. | ||
| ON | (5′ → 3′) | RNA target | DNA target | ||
|---|---|---|---|---|---|
| T m | ΔTm | T m | ΔTm | ||
| a = AM, p = PO, = 2′-OMe uridine. |
|||||
| ON6 | TaTaTaTaTpCpCpTpGpApTpApGpTaTaTaTaT | 54.6 | −1.5 | 52.3 | −3.2 |
| ON7 | pppppCpCpTpGpApTpApGppppp |
58.6 | +2.5 | 47.8 | −7.7 |
| ON8 | TpTpTpTpTpCpCpTpGpApTpApGpTpTpTpTpT | 56.1 | — | 55.5 | — |
No correlation was observed between the cleavage rate of an RNA target by RNase H with ON6–8 and the measured melting temperatures (Tms, Table 1 and Fig. S4, ESI†). The 2′-OMe modifications in gapmer ON7 increased target affinity to RNA by +2.5 °C (+0.25 °C per modification (mod)) whereas the amide backbone in ON6 slightly decreased duplex stability with RNA by −1.5 °C (−0.19 °C per mod) compared to unmodified ON8. This is consistent with the literature for both modifications.11,25
A flexible 4′-endo sugar pucker, a rigid sugar phosphate backbone, and a duplex conformation between A and B are essential for an efficient RNase H response.36 In this context, we investigated the structural changes in duplexes induced by the AM backbone. We did not observe significant perturbation of the duplex structures formed by ON6 with DNA or RNA targets when compared to ON8 (an unmodified DNA ON) in circular dichroism experiments (Fig. S5, ESI†), which is consistent with the high efficiency of ON6 in inducing an RNase H response.
Enzymatic stability of ASOs is important to ensure optimal biological half-life and therapeutic efficacy. The terminal amides in ON6 result in enhanced nuclease resistance and longer serum lifetime in foetal bovine serum (FBS) compared to ON7 and ON8 which were both rapidly degraded (Fig. S6, ESI†). As previously reported,37 the 2′-OMe modifications in ON7 slightly extend serum lifetime but are not as effective as the amide bonds in ON6.
Potential enhanced cellular uptake of the AM-gapmer design was evaluated by confocal laser scanning microscopy (CLSM). For this experiment, ON6 was fluorescently labelled at the 5′-end to give ON9 (Fig. 3D–F) in which eight PO linkages were replaced by charge-neutral AM bonds and the ON has a charge-to-linkage ratio of 0.56 (including the PO linkage between FL and the 5′-end). No solubility issues were encountered with this oligomer. ON10 and ON11 represent the fluorescently labelled derivatives of ON7 and ON8 respectively in which all 17 internucleoside linkages consist of negatively charged PO bonds, with an additional PO bond for the attachment of FL (Fig. 5). HeLa cells were incubated with 5 μM ON9–ON11 in serum-free medium and analysed by CLSM after fixation. AM-gapmer ON9 showed increased intracellular accumulation compared to ON10 and ON11 for which a distinct fluorescent signal inside the cell was absent (Fig. 5 and Fig. S7 for a general view, ESI†). The localised fluorescent signals observed for ON9 (Fig. 5A) suggest that the uptake mechanism leads to its partial entrapment within subcellular compartments. Co-incubation of ON9 with fluorescently tagged epidermal growth factor (EGF) shows that only a low number of FL signals were co-localised with stained endosomes (Fig. S8, ESI†). Moreover, a clear increase of punctuated fluorescence for ON9 was only detected after incubation for 16 h while receptor-mediated endocytosis is known to happen within <30 min. Together, our preliminary results suggest that ON uptake may be occurring through fluid phase endocytosis which results in unspecific uptake from the extracellular fluid into vacuoles, while their fate upon maturation can be highly variable.38 These observations are also in agreement with reports that fully charge-neutral methyl phosphonate-modified ASOs enter the cell via fluid phase endocytosis after 10–12 hours of incubation.21 Similar results showed enhanced cellular uptake of ASOs in which half the charge was neutralised by phosphotriesters.19
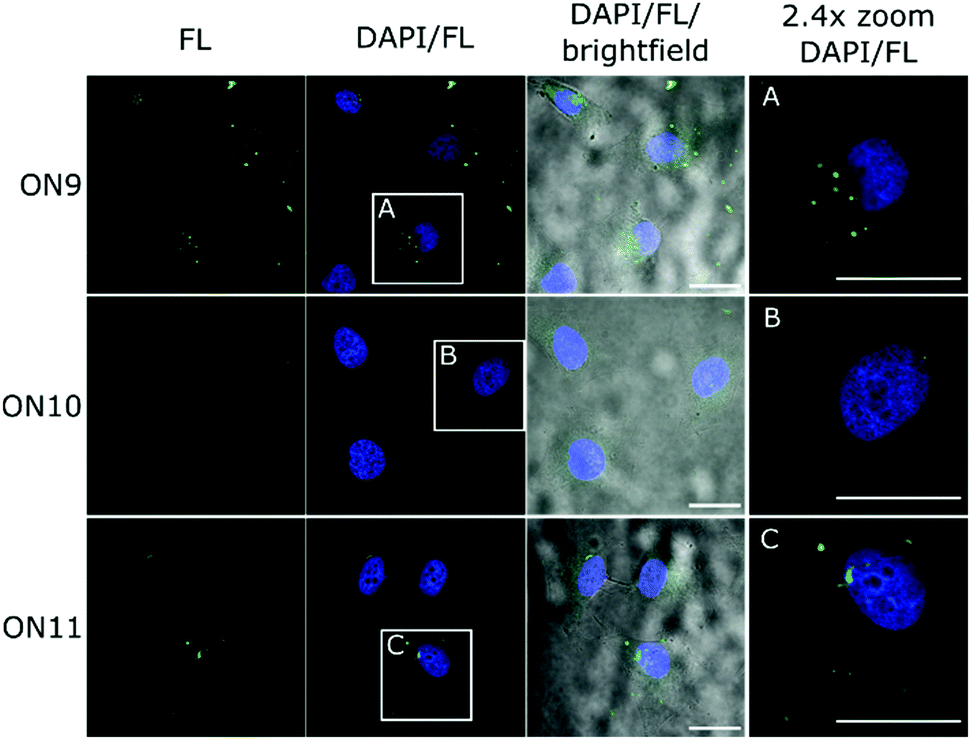 | ||
| Fig. 5 HeLa cells were incubated with 5 μM ON9–ON11 in serum-free medium for 16 h at 37 °C. Cells were fixed and nuclei stained with 4′,6-diamidino-2-phenylindole (DAPI) before CLSM analysis. Scale bar: 25 μm. ON9: 5′-FLpTaTaTaTaTpCpCpTpGpApTpApGpTaTaTaTaT-3′; ON10: 5′-FLppppppCpCpTpGpApTpApGppppp-3′; ON11: 5′-FLpTp TpTpTpTpCpCpTpGpApTpApGpTpTpTpTpT-3′. | ||
In conclusion, different ON chemistries and their effect on pharmacodynamics, pharmacokinetics, cellular delivery and toxicity are poorly understood and backbone modifications have been mainly focused on the PS linkage, leaving other chemistries underexplored. The recently reported applications of charge-neutral backbones to enhance cellular uptake19–21 and mediate toxicity22 emphasise the importance of exploring other artificial backbone structures. Here we report the partial replacement of the natural ON PO backbone by an amide internucleoside linkage and its effects on antisense activity, target engagement, serum stability and cellular uptake. We show that the AM linkage is well tolerated in the wings of an ASO gapmer design while retaining an efficient RNase H response. The stability of the formed duplex is only slightly decreased (−0.19 °C per mod) compared to unmodified DNA, and this does not compromise target engagement and RNA cleavage efficiency. The ease of chimeric gapmer design and synthesis with reduced steric complexity, retained catalytic efficiency, high serum stability and potentially enhanced cellular uptake all add desirable drug-like properties. Thus, the AM backbone mimic represents a valuable candidate for further development.
S. E. thanks EPSRC SBMCDT (EP/L015838/1) for a studentship, C. T. is supported by BBSRC grant BB/M011224/1, A. H. E.-S. by BBSRC grant BB/R008655/1 and YB by BBSRC grant BB/S018794/1.
Conflicts of interest
There are no conflicts to declare.Notes and references
- S. T. Crooke, Nucleic Acid Ther., 2017, 27, 70–77 CrossRef CAS PubMed.
- C. I. E. Smith and R. Zain, Annu. Rev. Pharmacol. Toxicol., 2019, 59, 605–630 CrossRef CAS PubMed.
- A. Khvorova and J. K. Watts, Nat. Biotechnol., 2017, 35, 238–248 CrossRef CAS PubMed.
- C. F. Bennett, Annu. Rev. Med., 2019, 70, 307–321 CrossRef CAS PubMed.
- S. J. Keam, Drugs, 2018, 78, 1371–1376 CrossRef CAS PubMed.
- S. M. Hoy, Drugs, 2018, 78, 1625–1631 CrossRef CAS PubMed.
- C. Rinaldi and M. J. A. Wood, Nat. Rev. Neurol., 2017, 14, 9–21 CrossRef PubMed.
- R. S. Geary, T. A. Watanabe, L. Truong, S. Freier, E. A. Lesnik, N. B. Sioufi, H. Sasmor, M. Manoharan and A. A. Levin, J. Pharmacol. Exp. Ther., 2001, 296, 890–897 CAS.
- R. S. Geary, D. Norris, R. Yu and C. F. Bennett, Adv. Drug Delivery Rev., 2015, 87, 46–51 CrossRef CAS PubMed.
- W. Brad Wan and P. P. Seth, J. Med. Chem., 2016, 59, 9645–9667 CrossRef PubMed.
- S. Freier and K.-H. Altmann, Nucleic Acids Res., 1997, 25, 4429–4443 CrossRef CAS PubMed.
- N. Iwamoto, D. C. D. Butler, N. Svrzikapa, S. Mohapatra, I. Zlatev, D. W. Y. Sah, Meena, S. M. Standley, G. Lu, L. H. Apponi, M. Frank-Kamenetsky, J. J. Zhang, C. Vargeese and G. L. Verdine, Nat. Biotechnol., 2017, 35, 845–851 CrossRef CAS PubMed.
- J. M. Campbell, T. A. Bacon and E. Wickstrom, J. Biochem. Biophys. Methods, 1990, 20, 259–267 CrossRef CAS PubMed.
- T. A. Watanabe, R. S. Geary and A. A. Levin, Oligonucleotides, 2006, 16, 169–180 CrossRef CAS PubMed.
- H. J. Gaus, R. Gupta, A. E. Chappell, M. E. Østergaard, E. E. Swayze and P. P. Seth, Nucleic Acids Res., 2019, 47, 1110–1122 CrossRef CAS PubMed.
- X. H. Liang, H. Sun, W. Shen and S. T. Crooke, Nucleic Acids Res., 2015, 43, 2927–2945 CrossRef CAS PubMed.
- T. A. Vickers, M. Rahdar, T. P. Prakash and S. T. Crooke, Nucleic Acids Res., 2019, 47, 10865–10880 CrossRef PubMed.
- S. T. Crooke and R. S. Geary, Br. J. Clin. Pharmacol., 2013, 76, 269–276 CrossRef CAS PubMed.
- D. Sheehan, B. Lunstad, C. M. Yamada, B. G. Stell, M. H. Caruthers and D. J. Dellinger, Nucleic Acids Res., 2003, 31, 4109–4118 CrossRef CAS PubMed.
- B. R. Meade, K. Gogoi, A. S. Hamil, C. Palm-Apergi, A. Van Den Berg, J. C. Hagopian, A. D. Springer, A. Eguchi, A. D. Kacsinta, C. F. Dowdy, A. Presente, P. Lönn, M. Kaulich, N. Yoshioka, E. Gros, X. S. Cui and S. F. Dowdy, Nat. Biotechnol., 2014, 32, 1256–1261 CrossRef CAS PubMed.
- Y. Shoji, S. Akhtar, A. Periasamy, B. Herman and R. L. Juliano, Nucleic Acids Res., 1991, 19, 5543–5550 CrossRef CAS PubMed.
- M. T. Migawa, W. Shen, W. B. Wan, G. Vasquez, M. E. Oestergaard, A. Low, C. L. De Hoyos, R. Gupta, S. Murray, M. Tanowitz, M. Bell, J. G. Nichols, H. Gaus, X. Liang, E. E. Swayze, S. T. Crooke and P. P. Seth, Nucleic Acids Res., 2019, 47, 5465–5479 CrossRef CAS PubMed.
- S. Miyatake, Y. Mizobe, M. K. Tsoumpra, K. R. Q. Lim, Y. Hara, F. Shabanpoor, T. Yokota, S. Takeda and Y. Aoki, Mol. Ther. – Nucleic Acids, 2019, 14, 520–535 CrossRef CAS PubMed.
- A. De Mesmaeker, A. Waldner, J. Lebreton, P. Hoffmann, V. Fritsch, R. M. Wolf and S. M. Freier, Angew. Chem., Int. Ed. Engl., 1994, 33, 226–229 CrossRef.
- P. von Matt, A. De Mesmaeker, U. Pieles, W. Zürcher and K. H. Altmann, Tetrahedron Lett., 1999, 40, 2899–2902 CrossRef CAS.
- A. Shivalingam, A. E. S. Tyburn, A. H. El-Sagheer and T. Brown, J. Am. Chem. Soc., 2017, 139, 1575–1583 CrossRef CAS PubMed.
- D. Mutisya, T. Hardcastle, S. K. Cheruiyot, P. S. Pallan, S. D. Kennedy, M. Egli, M. L. Kelley, A. van, B. Smith and E. Rozners, Nucleic Acids Res., 2017, 45, 8142–8155 CrossRef CAS PubMed.
- I. Van Daele, H. Munier-Lehmann, M. Froeyen, J. Balzarini and S. Van Calenbergh, J. Med. Chem., 2007, 50, 5281–5292 CrossRef CAS PubMed.
- M. Nowotny, S. A. Gaidamakov, R. Ghirlando, S. M. Cerritelli, R. J. Crouch and W. Yang, Mol. Cell, 2007, 28, 264–276 CrossRef CAS PubMed.
- B. M. Brandsen, A. R. Hesser, M. A. Castner, M. Chandra and S. K. Silverman, J. Am. Chem. Soc., 2013, 135, 16014–16017 CrossRef CAS PubMed.
- P. Tanui, S. D. Kennedy, B. D. Lunstad, A. Haas, D. Leake and E. Rozners, Org. Biomol. Chem., 2014, 12, 1207–1210 RSC.
- V. Kotikam, J. A. Viel and E. Rozners, Chem. – Eur. J., 2020, 26, 685–690 CrossRef CAS PubMed.
- B. P. Monia, E. A. Lesnik, C. Gonzalez, W. F. Lima, D. McGee, C. J. Guinosso, A. M. Kawasaki, P. D. Cook and S. M. Freier, J. Biol. Chem., 1993, 286, 14514–14522 Search PubMed.
- C. Malchére, J. Verheijen, S. Van der Laan, L. Bastide, J. Van Boom, B. Lebleu and I. Robbins, Antisense Nucleic Acid Drug Dev., 2011, 10, 463–468 CrossRef PubMed.
- A. J. Debacker, V. K. Sharma, P. Meda Krishnamurthy, D. O’Reilly, R. Greenhill and J. K. Watts, Biochemistry, 2019, 58, 582–589 CrossRef CAS PubMed.
- W. F. Lima, J. G. Nichols, H. Wu, T. P. Prakash, M. T. Migawa, T. K. Wyrzykiewicz, B. Bhat and S. T. Crooke, J. Biol. Chem., 2004, 279, 36317–36326 CrossRef CAS PubMed.
- P. Martin, Helv. Chim. Acta, 1995, 78, 486–504 CrossRef CAS.
- J. P. Lim and P. A. Gleeson, Immunol. Cell Biol., 2011, 89, 836–843 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0cc00444h |
| This journal is © The Royal Society of Chemistry 2020 |