
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAminocarboxylic acids related to aspergillomarasmine A (AMA) and ethylenediamine-N,N′-disuccinic acid (EDDS) are strong zinc-binders and inhibitors of the metallo-beta-lactamase NDM-1†
Kamaleddin H. M. E.
Tehrani‡
a,
Haigen
Fu‡
b,
Nora C.
Brüchle
 a,
Vida
Mashayekhi
c,
Alejandro
Prats Luján
b,
Matthijs J.
van Haren
a,
Gerrit J.
Poelarends
*b and
Nathaniel I.
Martin
*a
a,
Vida
Mashayekhi
c,
Alejandro
Prats Luján
b,
Matthijs J.
van Haren
a,
Gerrit J.
Poelarends
*b and
Nathaniel I.
Martin
*a
aBiological Chemistry Group, Institute of Biology Leiden, Leiden University, Sylviusweg 72, 2333 BE Leiden, The Netherlands. E-mail: n.i.martin@biology.leidenuniv.nl
bDepartment of Chemical and Pharmaceutical Biology, University of Groningen, Antonius Deusinglaan 1, 9713 AV Groningen, The Netherlands. E-mail: g.j.poelarends@rug.nl
cDivision of Cell Biology, Department of Biology, Faculty of Science, Utrecht University, Utrecht, The Netherlands
First published on 5th February 2020
Abstract
A series of aminocarboxylic acid analogues of aspergillomarasmine A (AMA) and ethylenediamine-N,N'-disuccinic acid (EDDS) were chemoenzymatically synthesized via the addition of various mono- and diamine substrates to fumaric acid catalyzed by the enzyme EDDS lyase. Many of these novel AMA and EDDS analogues demonstrate potent inhibition of the bacterial metallo-β-lactamase NDM-1. Isothermal titration calorimetry assays revealed a strong correlation between the inhibitory potency of the compounds and their ability to bind zinc. Compounds 1a (AMA), 1b (AMB), 5 (EDDS), followed by 1d and 8a, demonstrate the highest synergy with meropenem resensitizing an NDM-1 producing strain of E. coli to this important carbapenem of last resort.
Antibiotic resistance is a global public health concern with an increasing economic burden.1,2 Among Gram-negative pathogens, β-lactam resistance due to the production of β-lactamases is a major cause of antibiotic resistance.3 Based on their mechanism of β-lactam hydrolysis, β-lactamases can be classified as serine- or metallo-β-lactamases (SBLs and MBLs respectively). While SBLs hydrolyze β-lactams via an active site serine nucleophile, MBLs do so via a water molecule coordinated with active site zinc ion(s).4 Although there are clinically used SBL inhibitors available to counteract the infections caused by SBL-producing bacteria,5 there are currently no approved MBL inhibitors available.
Recent screening efforts led to the identification of aspergillomarasmine A (AMA, 1a, Table 1) as a potent inhibitor of the clinically relevant NDM- and VIM-type MBLs.6 This finding was followed by reports describing the chemical synthesis of AMA and its structural analogues.7–11 Among them, synthetic routes using either a key N-nosyl protected aziridine intermediate10 or a cyclic sufamidate9 furnished AMA in relatively few steps and the highest reported yields (overall yields of 28% and 19% respectively).
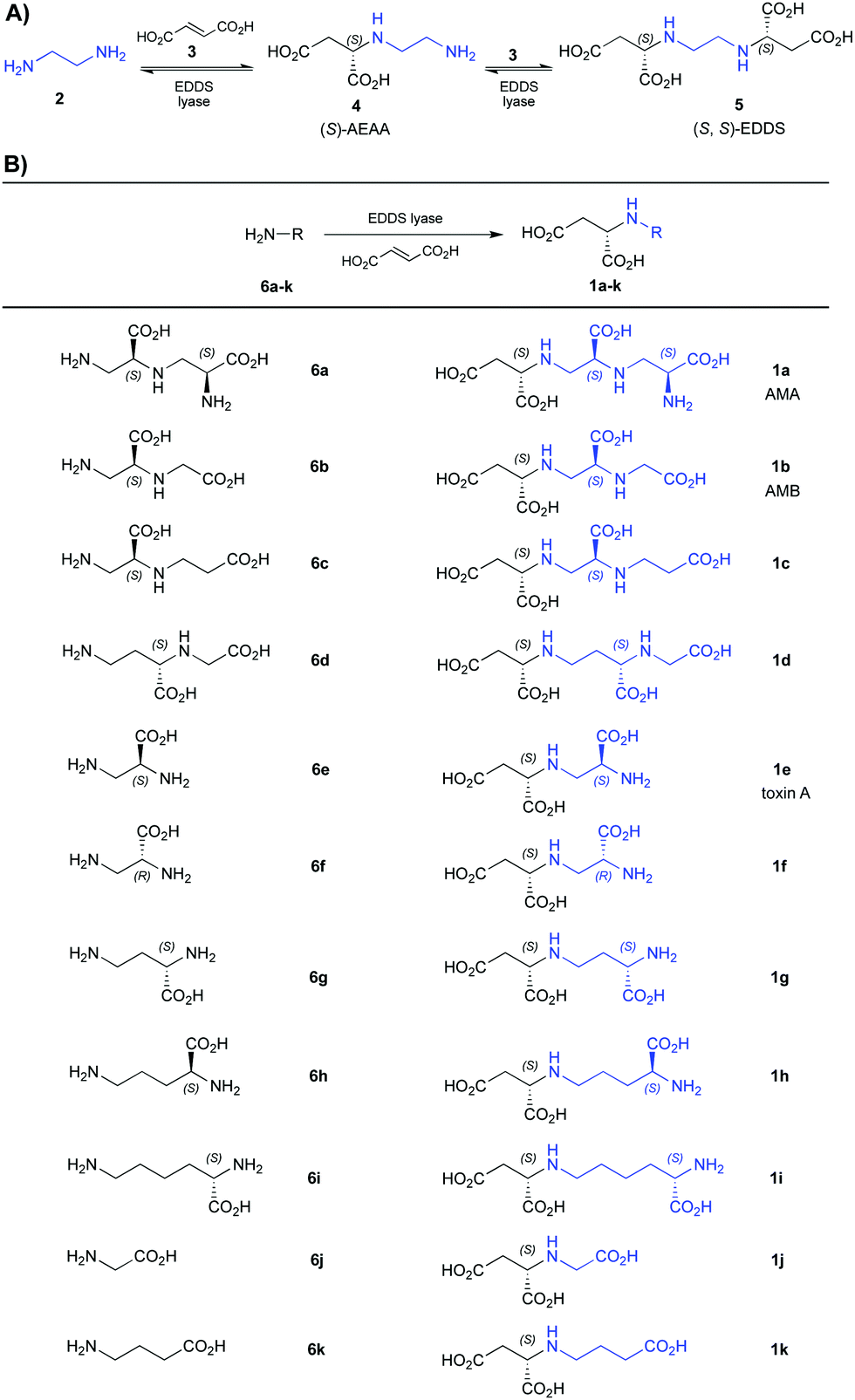
| (A) Natural reaction catalyzed by EDDS lyase. (B) Previously reported12,13 analogues of AMA, AMB, and toxin A prepared using the EDDS lyase methodology and here investigated as NDM-1 inhibitors. |
|---|
|
Recently, we reported that ethylenediamine-N,N′-disuccinic acid (EDDS) lyase naturally catalyzes a reversible two-step sequential addition of ethylenediamine (2) to two molecules of fumaric acid (3), giving (S)-N-(2-aminoethyl)aspartic acid (AEAA, 4) as an intermediate and (S,S)-EDDS (5) as the final product (Table 1A).12
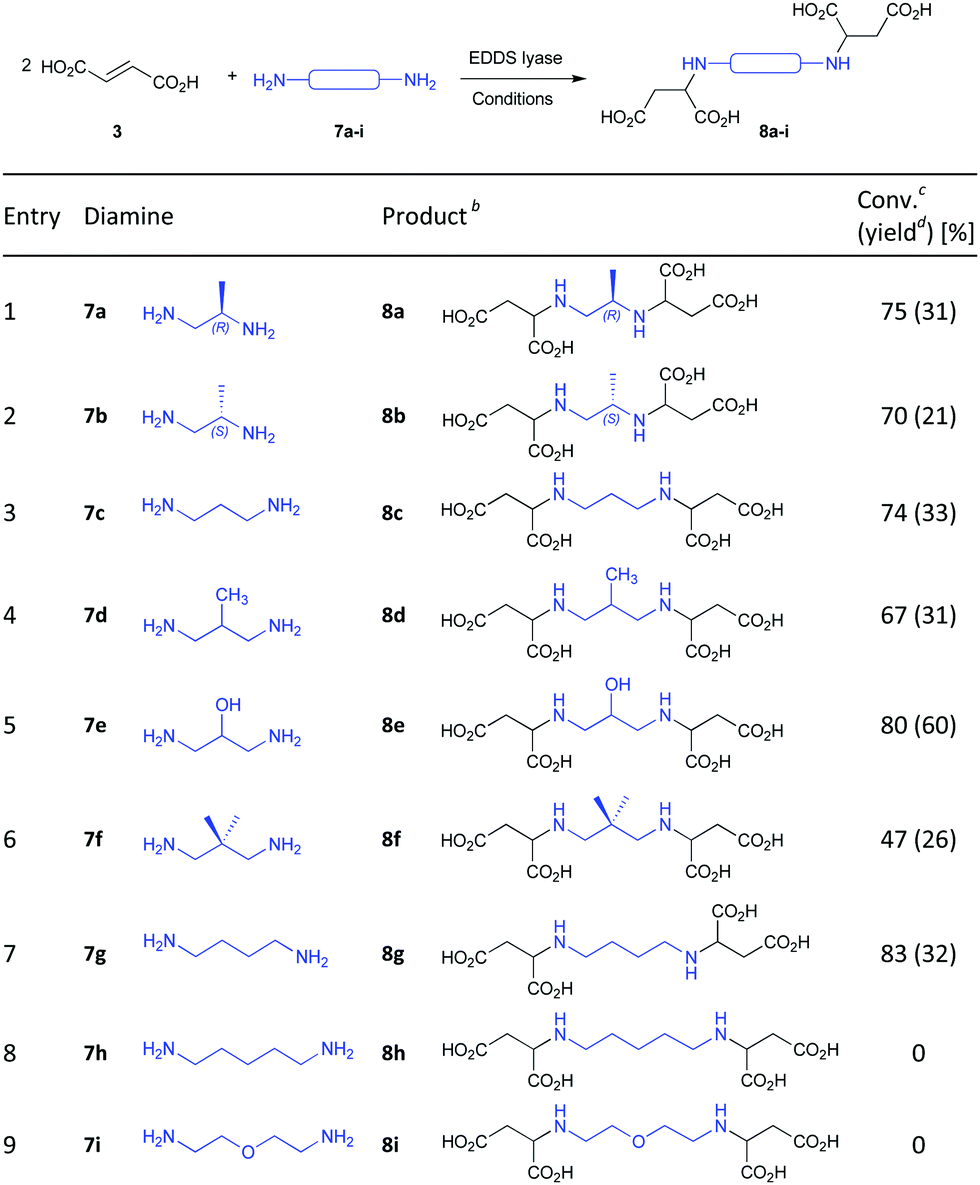
EDDS lyase was subsequently found to have broad substrate promiscuity,13–15 accepting a wide range of amino acids with terminal amino groups (6a–k) for regio- and stereoselective addition to fumarate, thus providing a straightforward biocatalytic method for the asymmetric synthesis of AMA (1a), AMB (1b), and related aminocarboxylic acids (1c–k, Table 1B).13 To further explore the substrate scope of EDDS lyase, as well as to prepare a small library of EDDS derivatives as potential NDM-1 inhibitors,16 we here describe the EDDS-lyase catalyzed reaction of fumaric acid with various diamines containing different aliphatic linkers between the two amino functional groups (7a–i) (Table 2). Interestingly, diamine substrates with two to four atoms between the two amino groups (7a–g) were well accepted as substrates by EDDS lyase, giving good conversions (47–83%) and yielding the corresponding aminocarboxylic acid products (8a–g) in 21–60% isolated yield (Table 2, entries 1–7). Hence, EDDS lyase has a broad diamine scope, allowing the two-step sequential addition of appropriate diamines to fumaric acid, providing a powerful synthetic tool for the preparation of valuable aminocarboxylic acids. Conversely, the elongated diamines with five atoms between the two amino groups (7h–i) were not accepted as substrates by EDDS lyase (Table 2, entries 8 and 9).
| a Conditions and reagents: reaction mixture (15 mL) consisted of fumaric acid (3, 60 mM), a diamine substrate (7a–i, 10 mM) and purified EDDS lyase (0.05 mol% based on diamine) in 50 mM Na2HPO4 buffer (pH 8.5). The reaction mixture was incubated at room temperature for 48 h (7a–e and 7g) or 96 h (7f and 7h–i). b Absolute stereochemistry of products not determined. c Conversion yields based on comparing 1H NMR signals of substrates and corresponding products. d Isolated yield after ion-exchange chromatography. |
|---|
|
The ability of the AMA and EDDS analogues to inhibit NDM-1 was evaluated using a fluorescence-based assay previously described by Schofield and coworkers.17 This assay makes use of a cephalosporin substrate known as FC5 which upon hydrolysis releases 7-hydroxycoumarin. The well characterized NDM-1 inhibitors AMA, EDTA, and dipicolinic acid (DPA) were used as positive controls. In general, most of the AMA and EDDS analogues tested showed potent activity against NDM-1 with IC50 values ranging from 1.3 μM to 18.3 μM (Table 3).
| Compound | IC50a (μM) | FICIb,c |
|---|---|---|
| a The half-maximal inhibitory concentration of the compounds tested against NDM-1 using FC5 as substrate. b FICI: fractional inhibitory concentration index. FICI < 0.5 indicates synergy (see main text for formula used to calculate FICI). c The test microorganism was E. coli RC0089, an NDM-1 positive patient isolate with an MIC for meropenem of 32 μg mL−1. | ||
| 1a (AMA) | 0.94 ± 0.11 | 0.063 |
| 1b (AMB) | 2.63 ± 0.10 | 0.063 |
| 1c | 1.35 ± 0.12 | 0.156 |
| 1d | 1.37 ± 0.04 | 0.094 |
| 1e | 2.33 ± 0.18 | >0.281 |
| 1f | 2.89 ± 0.24 | >0.281 |
| 1g | 18.34 ± 3.67 | >0.281 |
| 1h | >400 | >0.281 |
| 1i | >400 | >0.281 |
| 1j | 7.87 ± 0.29 | >0.281 |
| 1k | >400 | >0.281 |
| 5 (EDDS·3Na) | 2.21 ± 0.39 | 0.047 |
| 8a | 4.33 ± 0.11 | 0.094 |
| 8b | 9.65 ± 0.16 | 0.281 |
| 8c | 3.11 ± 0.19 | >0.281 |
| 8d | 2.85 ± 0.10 | >0.281 |
| 8e | 3.50 ± 0.16 | >0.281 |
| 8f | 2.85 ± 0.04 | 0.156 |
| 8g | >400 | >0.281 |
| EDTA·2Na | 1.25 ± 0.06 | 0.047 |
| DPA | 4.94 ± 0.22 | 0.094 |
Compared with its analogues 8a–g, EDDS (5) proved to possess the highest activity (IC50 = 2.21 μM). Modifications to the central aliphatic spacer in length or steric bulk (or both) were generally tolerated. However, elongation of the linker to four methylene units (8g) led to the complete loss of activity. The inhibitory activity of the naturally occurring AMB (1b) was also promising (IC50 = 2.63 μM). Insertion of a methylene group as in compounds 1c and 1d maintained the activity leading to equipotent new AMB analogues.
Toxin A (1e) is believed to be the biosynthetic precursor of the related fungal aminocarboxylic acid AMA and AMB.18 We found low IC50 values for toxin A (1e) and its diastereomer 1f (IC50 = 2.33 μM and 2.89 μM respectively). Replacing the diaminopropionic acid moiety with the much simpler glycine unit as in 1j led to a slight reduction of potency. Notably, elongation of the aliphatic spacers in both 1e and 1j, to generate compounds 1g–i and 1k, resulted in further or complete loss of NDM-1 inhibitory activity.
The majority of MBL inhibitors reported to date owe their activity to an ability to bind zinc. In general, MBL inhibitors either coordinate with zinc ions within the MBL active site or, if they are strong enough chelators, actively strip zinc from the MBL active site rendering the enzyme inactive.19,20 We have previously shown that the zinc-binding capacity of MBL inhibitors can be conveniently quantified using isothermal titration calorimetry (ITC).21 To this end, we next measured the zinc-binding affinity of the aminocarboxylic acids analogues listed in Table 3. These studies were conducted by titrating a zinc-containing solution into the test compound with the heat of binding monitored using a microcalorimeter (see supplementary information for more detail). The relevant thermodynamic parameters thus obtained (Kd and ΔH) are presented in supplemental Table S1. For compounds 1e–f, 1j, and 8c strong zinc binding was established with Kd values in the nM range. Notably, in the case of compounds 1b–d, 5, 8a, 8b, 8d, 8e, and 8f, the zinc binding interactions were found to be so strong (Kd < 100 nM) that only ΔH values could be accurately determined. By comparison, for 1h, 1i, 1k, and 8g the zinc binding was too weak to allow for a reliable determination of any thermodynamic parameters. The data thus obtained reveals a clear correlation between zinc-binding affinity and the IC50 values measured against NDM-1. These findings indicate that the major mechanism of NDM-1 inhibition for the aminocarboxylic acids here studied can be attributed to their ability to bind zinc.
The compounds were also tested for their ability to resensitize an NDM-1 producing E. coli isolate to meropenem, a clinically important carbapenem antibiotic. This NDM-1 expressing strain of E. coli was found to be highly resistant to carbapenem antibiotics with a minimum inhibitory concentration (MIC) of 32 μg mL−1 for meropenem. As a measure of potency, we determined the “rescue concentration” of each test compound (see ESI,† Table S2) which provides an indication of synergy.9 Rescue concentration is defined as the lowest concentration of an MBL inhibitor that can resensitize a resistant strain to the antibiotic of interest when applied at its clinical breakpoint concentration (1 μg mL−1 for meropenem). In addition, the fractional inhibitory concentration index (FICI) values were determined for each compound and are provided in Table 3. FICI values were established by applying the following formula where an FICI < 0.5 indicates synergy:
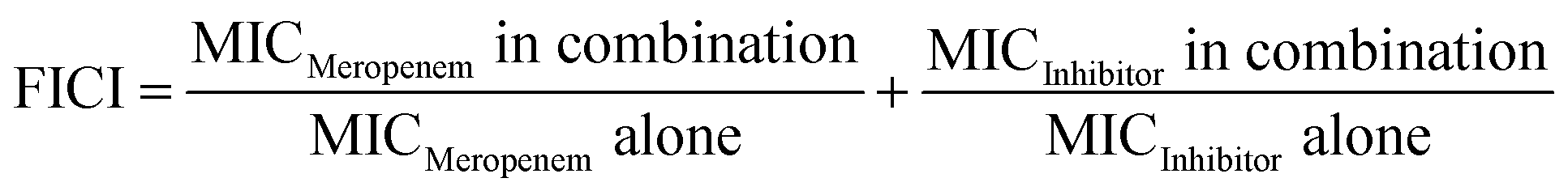
Among the EDDS analogues examined, 5 followed by 8a were among the most potent synergizers. AMB (1b) and its related analogues 1c and 1d also showed potent to moderate activity. Interestingly, neither toxin A or its analogues (1e–k) demonstrated potent synergistic activity suggesting they may not be able to effectively access the enzyme target in the microorganism.
In conclusion, we here describe the application of a robust chemoenzymatic synthesis route in the preparation of a series of novel aminocarboxylic acids. A number of these compounds were found to be potent inhibitors of NDM-1, with inhibitory activities well correlated to zinc binding ability. In addition, a number of the most active compounds demonstrated promising synergistic activity against an NDM-1 producing E. coli when combined with meropenem. In the search for new agents to combat antibiotic resistance, chemoenzymatic methodologies such as those here described have the potential to provide access to novel inhibitors of metallo-β-lactamases of clinical relevance.
Professor Gerry Wright is kindly acknowledged for generously providing an authentic sample of aspergillomarasmine A. Financial support provided by The China Scholarship Council (PhD scholarship to HF) The European Research Council (ERC consolidator grant to NIM, grant agreement no. 725523).
Conflicts of interest
There are no conflicts to declare.Notes and references
- M. Ferri, E. Ranucci, P. Romagnoli and V. Giaccone, Crit. Rev. Food Sci. Nutr., 2017, 57, 2857–2876 CrossRef CAS.
- B. Aslam, W. Wang, M. I. Arshad, M. Khurshid, S. Muzammil, M. H. Rasool, M. A. Nisar, R. F. Alvi, M. A. Aslam, M. U. Qamar, M. K. F. Salamat and Z. Baloch, Infect. Drug Resist., 2018, 1645–1658 CrossRef CAS.
- A. K. Thabit, J. L. Crandon and D. P. Nicolau, Expert Opin. Pharmacother., 2015, 16, 159–177 CrossRef CAS PubMed.
- S. M. Drawz and R. A. Bonomo, Clin. Microbiol. Rev., 2010, 23, 160–201 CrossRef CAS.
- K. H. M. E. Tehrani and N. I. Martin, MedChemComm, 2018, 9, 1439–1456 RSC.
- A. M. King, S. A. Reid-Yu, W. Wang, D. T. King, G. De Pascale, N. C. Strynadka, T. R. Walsh, B. K. Coombes and G. D. Wright, Nature, 2014, 510, 503–506 CrossRef CAS.
- D. Liao, S. Yang, J. Wang, J. Zhang, B. Hong, F. Wu and X. Lei, Angew. Chem., Int. Ed., 2016, 55, 4291–4295 CrossRef CAS.
- K. Koteva, A. M. King, A. Capretta and G. D. Wright, Angew. Chem., Int. Ed., 2016, 55, 2210–2212 CrossRef CAS PubMed.
- S. A. Albu, K. Koteva, A. M. King, S. Al-Karmi, G. D. Wright and A. Capretta, Angew. Chem., Int. Ed., 2016, 55, 13259–13262 CrossRef CAS PubMed.
- J. Zhang, S. Wang, Y. Bai, Q. Guo, J. Zhou and X. Lei, J. Org. Chem., 2017, 82, 13643–13648 CrossRef CAS PubMed.
- J. Zhang, S. Wang, Q. Wei, Q. Guo, Y. Bai, S. Yang, F. Song, L. Zhang and X. Lei, Bioorg. Med. Chem., 2017, 25, 5133–5141 CrossRef CAS PubMed.
- H. Poddar, J. de Villiers, J. Zhang, V. Puthan Veetil, H. Raj, A.-M. W. H. Thunnissen and G. J. Poelarends, Biochemistry, 2018, 57, 3752–3763 CrossRef CAS PubMed.
- H. Fu, J. Zhang, M. Saifuddin, G. Cruiming, P. G. Tepper and G. J. Poelarends, Nat. Catal., 2018, 1, 186–191 CrossRef CAS.
- H. Fu, A. Prats Luján, L. Bothof, J. Zhang, P. G. Tepper and G. J. Poelarends, ACS Catal., 2019, 9, 7292–7299 CrossRef CAS.
- J. Zhang, H. Fu, P. G. Tepper and G. J. Poelarends, Adv. Synth. Catal., 2019, 361, 2433–2437 CrossRef CAS.
- A. Proschak, J. Kramer, E. Proschak and T. A. Wichelhaus, J. Antimicrob. Chemother., 2017, 73, 425–430 CrossRef PubMed.
- S. S. van Berkel, J. Brem, A. M. Rydzik, R. Salimraj, R. Cain, A. Verma, R. J. Owens, C. W. G. Fishwick, J. Spencer and C. J. Schofield, J. Med. Chem., 2013, 56, 6945–6953 CrossRef CAS PubMed.
- P. Friis, C. E. Olsen and B. L. Møller, J. Biol. Chem., 1991, 266, 13329–13335 CAS.
- L. C. Ju, Z. Cheng, W. Fast, R. A. Bonomo and M. W. Crowder, Trends Pharmacol. Sci., 2018, 39, 635–647 CrossRef CAS PubMed.
- C. M. Rotondo and G. D. Wright, Curr. Opin. Microbiol., 2017, 39, 96–105 CrossRef CAS PubMed.
- K. H. M. E. Tehrani and N. I. Martin, ACS Infect. Dis., 2017, 3, 711–717 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0cc00356e |
| ‡ Denotes equal contribution. |
| This journal is © The Royal Society of Chemistry 2020 |