
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceStructural elucidation of the degradation mechanism of nickel-rich layered cathodes during high-voltage cycling†
Jing
Lai
a,
Jun
Zhang
b,
Zuowei
Li
a,
Yao
Xiao
a,
Weibo
Hua
 *c,
Zhenguo
Wu
*a,
Yanxiao
Chen
a,
Yanjun
Zhong
a,
Wei
Xiang
d and
Xiaodong
Guo
a
*c,
Zhenguo
Wu
*a,
Yanxiao
Chen
a,
Yanjun
Zhong
a,
Wei
Xiang
d and
Xiaodong
Guo
a
aSchool of Chemical Engineering, Sichuan University, Chengdu 610065, P. R. China. E-mail: zhenguowu@scu.edu.cn
bRuyuan Dongyangguang Magnetic Materials Co., Ltd, Guangdong, Ruyuan 512000, P. R. China
cInstitute for Applied Materials (IAM), Karlsruhe Institute of Technology (KIT), Hermann-von-Helmholtz-Platz 1, 76344 Eggenstein-Leopoldshafen, Germany. E-mail: weibo.hua@kit.edu
dCollege of Materials and Chemistry & Chemical Engineering, Chengdu University of Technology, Chengdu, 610059, P. R. China
First published on 23rd March 2020
Abstract
Phase transition occurring during cycling plays a fundamentally important role in the cycling performance of nickel-rich cathodes. Here, splitting of two O3 phases, rather than the often observed O1 phases in the conventional LiCoO2 electrode, was discovered in LiNi0.85Co0.10Mn0.05O2 at a high-voltage region (>4.6 V). Such degradation could be mitigated via Al doping.
Lithium ion batteries (LIBs) are considered one of the most promising energy storage devices for battery electric vehicles (BEVs). Among the available cathode materials for LIBs, nickel-rich layered LiNixCoyM1−x−yO2 (x ≥ 0.8, M = Mn, Al, Fe, Mg, etc.) oxides are the ideal choice due to their higher capacity, lower cost and less environmental impact.1–4 However, nickel-rich layered oxide materials usually suffer from increasingly severe capacity fading, with increasing nickel content.5 Severe electrochemical performance degradation largely originates from sharp phase transitions, causing heterogeneous lattice expansion and contraction during electrochemical cycling.6 H2–H3 phase transition, in a deeply delithiated state, plays a key role in determining the structural stability of a nickel-rich layered cathode, as the transformation leads to detrimentally anisotropic volume change and aggravating capacity fading.
Up to now, it remains an open question whether the degradation mechanism of Ni-rich layered cathodes is a phase transition process or a solid-solution reaction during high-voltage cycling. For example, Manthiram7et al. reported the formation of a solid solution phase in LiNi0.92Co0.06Al0.02O2 electrodes during cycling, while Biasi8et al. demonstrated that the LiNiO2 cathode suffers from the phase transformation from a layered hexagonal H1 phase to a layered H2/H3 phase during the de-lithiation process. Recently, Sun's group9 reported that three phase transitions, i.e. H1 → H2, H2 → H3 and H3 → O1, are detected in LiNi0.95Co0.025Mn0.025O2, which means that the stacking sequence of oxygen atoms changes from cubic close-packing (ABCABC) in the H1/H2/H3 phase to hexagonal close-packing (ABAB) in the O1 phase when almost all Li-ions are extracted from the Li layer in the layered structure.
Here, in situ high-resolution synchrotron radiation diffraction (SRD) is used to investigate the structural evolution of a LiNi0.85Co0.10Mn0.05O2 cathode material (N85CM). Interestingly, new phase transitions were observed, which are different from those published in the literature.10–13 Two layered phases, i.e. H3a and H3b phases, with the same oxygen stacking (O3 structure) but different lattice parameters are generated after charging to above 4.6 V. To mitigate the degradation of N85CM during high-voltage cycling, small-dose of Al (2 mol%) for Mn in LiNi0.85Co0.10Mn0.03Al0.02O2 (N85CMA) is adopted. The in situ SRD results suggest that N85CMA experienced a solid-solution reaction within a wide voltage window (2.7–4.8 V vs. Li/Li+). Al-doping significantly suppresses the split hexagonal phase, proposed to mitigate the structural and functional fatigue of layered Ni-rich cathode materials during high-voltage cycling.
N85CM and N85CMA are synthesized by a sol–gel method followed by a conventional high-temperature solid-state method.14 For comparison, the traditional LiCoO2 cathode was also prepared. High-resolution synchrotron radiation diffraction (SRD) was performed (Fig. S1 in the ESI†) to investigate the structures of N85CM, LiCoO2 and N85CMA. All the diffraction reflections of the three samples can be indexed to a hexagonal α-NaFeO2 layered structure with a space group R-![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m. No obvious impurities can be observed. The electrochemical properties of the selected samples were tested at a cut-off voltage of 4.8 V, and the initial charge–discharge curves of the electrodes are shown in Fig. 1 and Fig. S4 (ESI†). Both N85CM and N85CMA electrodes are found to exhibit similar monotonous charge/discharge features.
m. No obvious impurities can be observed. The electrochemical properties of the selected samples were tested at a cut-off voltage of 4.8 V, and the initial charge–discharge curves of the electrodes are shown in Fig. 1 and Fig. S4 (ESI†). Both N85CM and N85CMA electrodes are found to exhibit similar monotonous charge/discharge features.
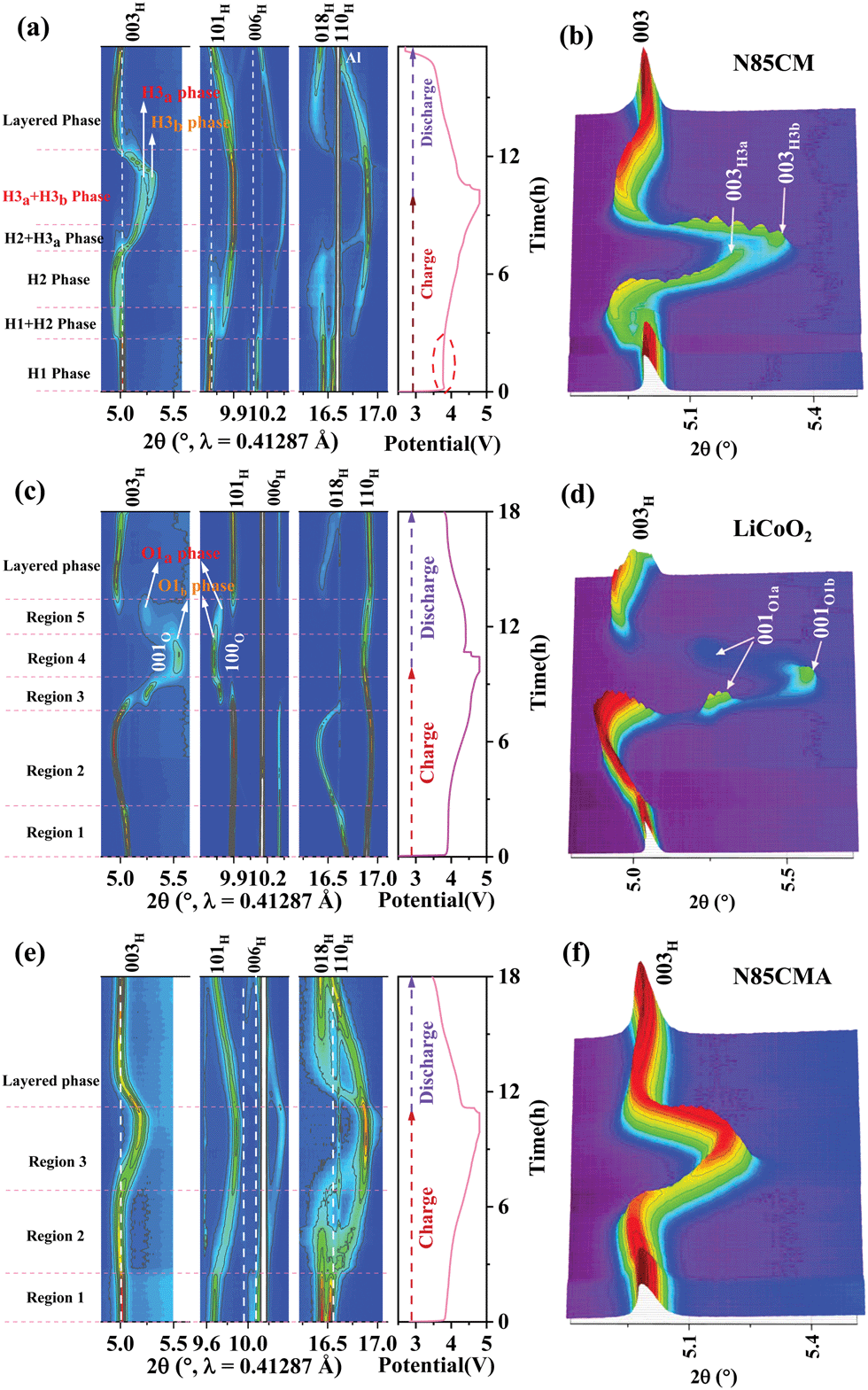 | ||
| Fig. 1 In situ SRD of (a) N85CM, (c) LiCoO2 and (e) N85CMA electrodes: Contour plot of reflection evolution of 003, 101, 006, 018 and 110 during the first charging and discharging process. The original SRD patterns of 003 reflection of (b) N85CM, (d) LiCoO2 and (f) N85CMA during the first cycle between 2.7 and 4.8 V. H and O indicate the hexagonal layered phase with an O3 structure and the trigonal layered phase with an O1 structure, respectively. | ||
To illuminate the structural evolution of N85CM during high-voltage cycling, in situ high-resolution SRD was performed in the voltage range of 2.7–4.8 V. The contour plots of intensity and positions for several reflections, i.e. 003, 101, 104, 018 and 110, are exhibited in Fig. 1a. The original SRD patterns of the 003 peak during the first cycle from 2.7–4.8 V of N85CM are exhibited in Fig. 1b, and the corresponding voltage profile of the first cycle at 2.7–4.8 V is illustrated in Fig. 1a. The scattering angles of 003, 104, and 018 reflections imply the change in the average metal–metal inter-slab distance of the layered structure (i.e. c lattice parameter), meanwhile 101 and 110 reflections represent the evolution of average metal–metal intra-slab distance (i.e. a lattice parameter).15 Interestingly, the galvanostatic curve of N85CM exhibits a steep increase of the voltage up to 3.88 V in the early stage of the charge, and then the voltage decreases to yield a plateau at 3.77 V (marked by a red dotted cycle). Such an increase and then a decrease of the overpotential may be caused by the residual lithium on the surface of the materials,16 because there is no obvious change in the reflections on the SRD patterns (see Fig. 1a and b). Upon charging to 3.8 V, a new set of 003, 101, 104, 018, and 110 reflections occurs, indicating the appearance of a new H2 phase.17,18 Afterwards, the H2 phase remains alone until 4.3 V upon charging. With further de-lithiation, a group of new reflections emerges with larger deviation from previous ones, which is defined as the H3a phase. Then, the H3a phase grows at the expense of the H2 phase and the co-existence remains until 4.3–4.5 V charge. Impressively, above 4.6 V, the characteristic reflections split again, implying a new phase with the Rm space group. The detailed SRD patterns are shown in Fig. 1b. The new phase with a larger deviation is identified as the H3b phase. The coexistence of H3a and H3b phases remains until final charging to 4.8 V. During lithiation, the 003, 104, and 018 reflections return to lower angles. From the result above, it can be concluded that N85CM suffers from severe structural degradation during high-voltage cycling, resulting in a large hysteresis between charge and discharge processes in N85CM (e.g. the sharply decreased voltage in the beginning of the discharging process in Fig. 1a).
Each in situ SRD pattern during charging–discharging cycles has been refined by Rietveld refinement with the Rm structure model, and the corresponding lattice parameters are shown in Fig. 2a. For N85CM, at an open-circuit voltage of 3.8 V, parameter a of phase H1 decreases from 2.875(1) to 2.872(1) Å, while parameter c slightly increases from 14.203(1) to 14.209(1) Å. The contraction of parameter a is supposed to be caused by the reducing radii of TM cations with higher valence states.19 The expansion of parameter c is believed to be due to the increased electrostatic repulsive force between the oxygen layer and the TM layer as a result of the decreased number of Li ions in the Li layer in N85CM.20,21 From 3.8 to 4.0 V, the lattice parameter a of both H1 and H2 phases gradually decreased, the c constant of the two phases increases firstly and then decreases, and the maximum values are 14.242(1) Å and 14.474(1) Å respectively, see Fig. 2a. Compared to the H1 phase, a higher c/a ratio indicates a better-defined crystal structure in the H2 phase.22 When charged to 4.3 V, parameter a of the H3a phase demonstrates a slight change and parameter c rapidly decreases from 14.039(1) to 13.765(1) Å. From 4.6 to 4.8 V, the lattice parameter a of H3a and H3b phases remains constant within the range of experimental error, while parameter c of the two layered phases separately shrinks from 13.747(1) to 13.728(1) Å and from 13.584(1) to 13.526(1) Å. This decrease in parameter c may have resulted from oxygen release and/or TM migration, leading to a reduction of the electrostatic repulsion between oxygen–oxygen layers along the c-axis and an expansion along the a (and b) axis. The lattice parameters (a, b, and c) of the H3b phase are always smaller than those of the H3a phase, leading to a larger change in mechanical strain among various phases and thus to undesired fracture and pulverization of materials, see Fig. 3. After discharge, both parameters a and c do not reach the original value of the pristine material before cycling and Δc and Δa are 0.0609(1) Å and −0.0032(1) Å respectively, demonstrating that the atomic structure of N85CM is not totally reversible, which may result from the incomplete insertion of lithium ions into the layered structure.23
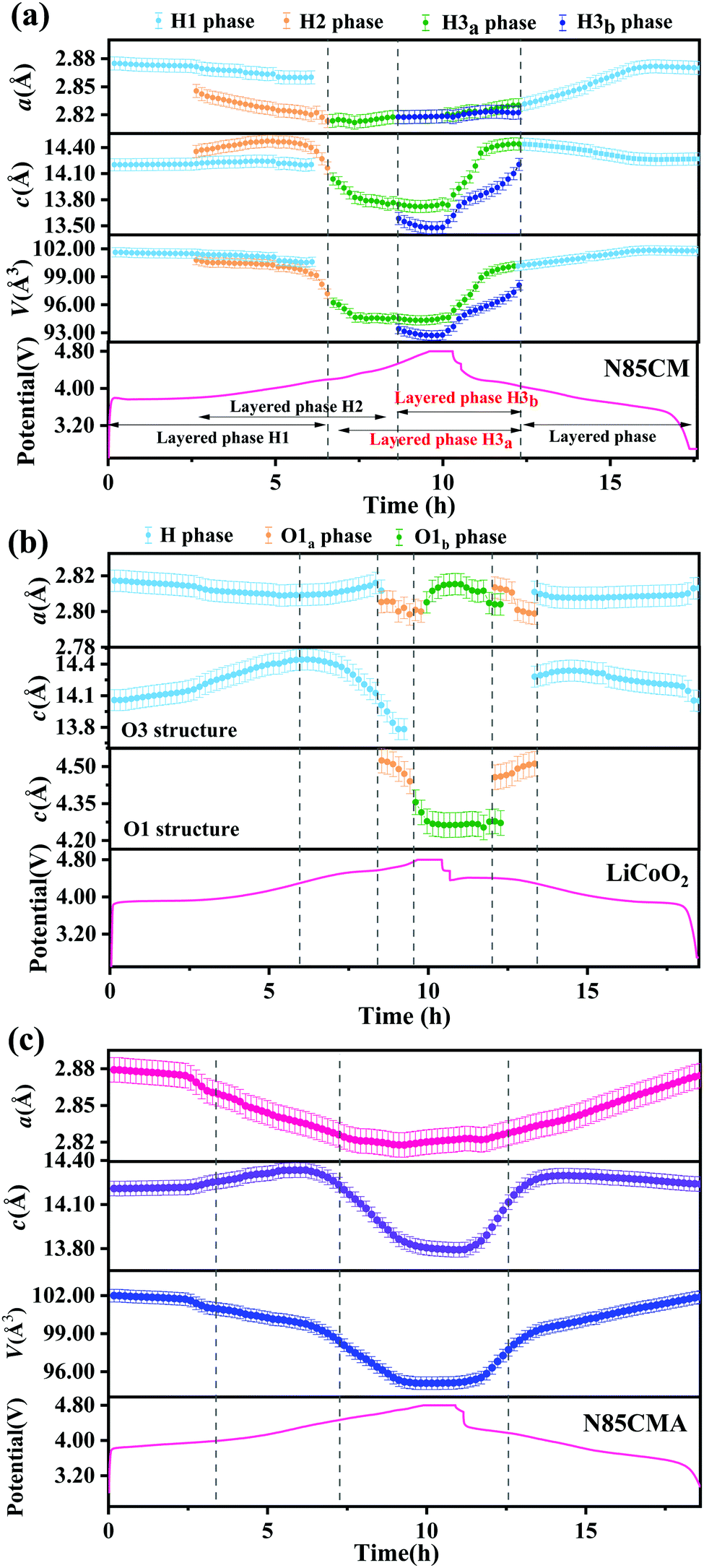 | ||
| Fig. 2 Lattice parameter changes of (a) N85CM, (b) LiCoO2 and (c) N85CMA cathodes during the first charge–discharge process. | ||
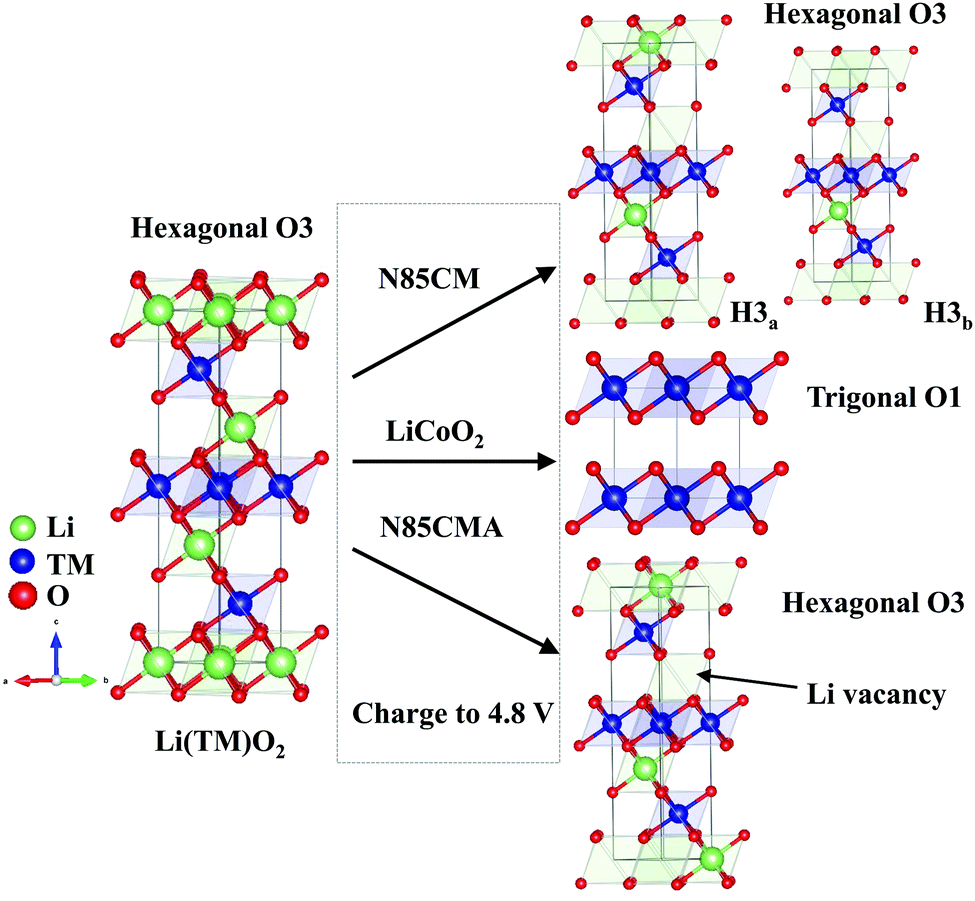 | ||
| Fig. 3 Schematic diagram of phase transition during de-intercalation from O3-Li(TM)O2 to O3-Li1−x(TM)O2 or O1-Li1−xCoO2 (x > 0.8). | ||
In order to clarify whether O3 oxygen stacking in the new layered H3b phase is maintained, in situ SRD was performed on the traditional LiCoO2 cathode during high-voltage cycling (shown in Fig. 1(c and d)). At a highly delithiated state (4.5–4.8 V), a set of reflections such as 001 and 100 indexing to trigonal O1-type Li1−xCoO2 (x > 0.8) is clearly probed. The observed O1a and O1b phases are in good agreement with the previous in situ XRD results of LiCoO2 reported in the literature,24 which are different with the phase transition mechanisms in N85CM. The evolution of the lattice parameters of the LiCoO2 cathode as a function of reaction time obtained from Rietveld refinement is shown in Fig. 2b, showing massive structural collapse in the c-axis upon high-voltage cycling.
In order to alleviate the severe structural deterioration of the N85CM material, we adopted a simple method of aluminum doping to synthesize N85CMA. Fig. 1c and Fig. S4b (ESI†) show the charge–discharge curve of N85CMA during the first cycle. The voltage of N85CMA increases steeply up to about 3.68 V and then reaches a plateau. No pronounced increase in the voltage polarization is found in N85CMA, which matches well with those observed in the literature.16 Within a wide voltage window (2.7–4.8 V vs. Li/Li+) and at 1.0C rate, N85CM delivers a discharge capacity of 165.8 mA h g−1 and 57.2% capacity retention after 70 cycles, while N85CMA delivers a discharge capacity of 182.2 mA h g−1 and 73.1% capacity retention after 70 cycles (Fig. S4c, ESI†). After cycles, compared with N85CMA, the surface of N85CM particles suffered from severe pulverization, demonstrated in Fig. S6 (ESI†). Therefore, the substitution of Al could remarkably enhance the long-term structural and chemical stability of Ni-rich layered materials, thus favoring an improved capacity of N85CMA during high-voltage cycling.
In situ high-resolution SRD measurements were performed in the voltage range of 2.7–4.8 V to study the structural evolution of N85CMA, as shown in Fig. 1e. The characteristic reflections 003, 101, 018 and 110 shift to lower angles firstly and then to higher angles with further de-lithiation. Besides, there is no clear reflection splitting during charging from 2.7 V to 4.8 V, indicating no pronounced biphasic region.7 The continuous and steady evolution of reflections of N85CMA indicates no clear structural phase transition with cycling, suggesting that N85CMA is solid solution with the homogeneous reaction in the process of charging and discharging.7,25 Furthermore, the profiles of the corresponding lattice parameters of N85CMA are shown in Fig. 2c. The curves of lattice parameters a and c are smooth and there is no new branch of stacking axis constants. A solid solution reaction process alleviates the mechanical and structural degradation, thus resulting in a superior electrochemical performance (see Fig. S4, ESI†), as also demonstrated in previous research.26–31
In summary, the structural evolution of layered Ni-rich cathode materials is investigated by high-resolution SRD. According to the results of in situ SRD, the N85CM cathode experiences complex phase transformations and a dramatically new phase transformation (H3a to H3b transition). The phase transformation causes a severe change in mechanical stress among various phases, triggering off the irreversible structural degradation and functional fatigue. It is found that the substitution of Al could effectively suppress the H2–H3 phase transformation, beneficial to the improvement of the electrochemical performance of Ni-rich cathode materials. With this new exploration and effective modification, more nickel rich cathode materials with excellent structural stability and electrochemical performance could be expected in the near future.
This work was supported by the National Natural Science Foundation of China (No. 21805198, 21878195, 21805018), the Youth Foundation of Sichuan University (No. 2017SCU04a08), the National Key Research and Development of China (grant no. 2016YFD0200404), and the Research Foundation for the Postdoctoral Program of Sichuan University (No. 2017SCU12018 and 2018SCU12045). W. H. received financial support from the Helmholtz – OCPC Postdoc-Program. Parts of this research were carried out at ALBA Light Source using MSPD beamline. We thank Dr Zhuo Zheng for help with data analysis. This work contributes to the research performed at CELEST (Center for Electrochemical Energy Storage Ulm-Karlsruhe).
Conflicts of interest
There are no conflicts to declare.Notes and references
- W. Hua, S. Wang, M. Knapp, S. J. Leake, A. Senyshyn, M. Yavuz, J. R. Binder, C. P. Grey, H. Ehrenberg, S. Indris and B. Schwarz, Nat. Commun., 2019, 24, 1–11 Search PubMed.
- J. Y. Liao and A. Manthiram, J. Power Sources, 2015, 282, 429–436 CrossRef CAS.
- W. Dong, X. X. Zeng, X. D. Zhang, J. Y. Li, J. L. Shi, Y. Xiao, Y. Shi, R. Wen, Y. X. Yin, T. S. Wang, C. R. Wang and Y. G. Guo, ACS Appl. Mater. Interfaces, 2018, 10, 18005–18011 CrossRef CAS PubMed.
- Y. Xiao, P. F. Wang, Y. X. Yin, Y. F. Zhu, Y. B. Niu, X. D. Zhang, J. Zhang, X. Yu, X. D. Guo, B. H. Zhong and Y. G. Guo, Adv. Mater., 2018, 30, 1803765 CrossRef PubMed.
- X. Q. Yang, X. Sun and J. McBreen, Electrochem. Commun., 1999, 1, 227–232 CrossRef.
- U. H. Kim, H. H. Ryu, J. H. Kim, R. Mucke, P. Kaghazchi and Y. K. Sun, Adv. Energy Mater., 2019, 9, 18 Search PubMed.
- J. Y. Li, W. D. Li, S. Y. Wang, K. Jarvis, J. H. Yang and A. Manthiram, Chem. Mater., 2018, 30, 3101–3109 CrossRef CAS.
- L. D. Biasi, A. Schiele, R.-A. Maria, G. Garcia, T. Brezesinski, P. Hartmann and J. Janek, ChemSusChem, 2019, 12, 2240 CrossRef PubMed.
- U.-H. Kim, H.-H. Ryu, J.-H. Kim, R. Mücke, P. Kaghazchi, C. S. Yoon and Y.-K. Sun, Adv. Energy Mater., 2019, 1803902 CrossRef.
- T. Chen, X. Li, H. Wang, X. X. Yan, L. Wang, B. W. Deng, W. J. Ge and M. Z. Qu, J. Power Sources, 2018, 374, 1–11 CrossRef CAS.
- T. Li, X. H. Li, Z. X. Wang and H. J. Guo, J. Power Sources, 2017, 342, 495–503 CrossRef CAS.
- A. Manthiram, J. C. Knight, S. T. Myung, S. M. Oh and Y. K. Sun, Adv. Energy Mater., 2016, 6, 1 Search PubMed.
- W. Liu, P. Oh, X. Liu, M. J. Lee, W. Cho, S. J. Chae, Y. Kim and J. Cho, Angew. Chem., Int. Ed., 2015, 54, 4440–4457 CrossRef CAS PubMed.
- S. W. Lee, H. Kim, M. S. Kim, H. C. Youn, K. Kang, B. W. Cho, K. C. Roh and K. B. Kim, J. Power Sources, 2016, 315, 261–268 CrossRef CAS.
- W. Hua, B. Schwarz, M. Knapp, A. Senyshyn, A. Missiul, X. K. Mu, S. N. Wang, C. Kubel, J. R. Binder, S. Indris and H. Ehrenberg, J. Electrochem. Soc., 2019, 166, A5025–A5032 CrossRef CAS.
- Y. You, H. Celio, J. Li, A. Dolocan and A. Manthiram, Angew. Chem., Int. Ed., 2018, 57, 6480 CrossRef CAS PubMed.
- Y. Xia, J. M. Zheng, C. Wang and M. M. Gu, Nano Energy, 2018, 49, 434–452 CrossRef CAS.
- J. L. Shi, D. D. Xiao, M. Ge, X. Yu, Y. Chu, X. Huang, X. D. Zhang, Y. X. Yin, X. Q. Yang, Y. G. Guo, L. Gu and L. J. Wan, Adv. Mater., 2018, 30, 1705575 CrossRef PubMed.
- Y. W. Tsai, B. J. Hwang, G. Ceder, H. S. Shen, D. G. Liu and J. F. Lee, Chem. Mater., 2005, 17, 3191–3199 CrossRef CAS.
- J. Li, E. L. Downie, L. Ma, W. D. Qiu and J. R. Dahn, J. Electrochem. Soc., 2015, 162, A1401–A1408 CrossRef CAS.
- Y. Xiao, Y. F. Zhu, H. R. Yao, P. F. Wang, X. D. Zhang, H. L. Li, X. N. Yang, L. Gu, Y. C. Li, T. Wang, Y. X. Yin, X. D. Guo, B. H. Zhong and Y. G. Guo, Adv. Energy Mater., 2019, 9, 1803978 CrossRef.
- J. Li, N. Zhang, H. Y. Li, A. Liu, Y. Q. Wang, S. Yin, H. H. Wu and J. R. Dahn, J. Electrochem. Soc., 2018, 165, A3544–A3557 CrossRef CAS.
- A. O. Kondrakov, A. Schimidt, J. Xu, H. Geßwein, R. Mönig, P. Hartmann, H. Sommer, T. Brezesinski and J. Janek, J. Phys. Chem. C, 2017, 121, 3286–3294 CrossRef CAS.
- K. Y. Chung, W. S. Yoon, H. S. Lee, J. McBreen, X. Q. Yang, S. H. Oh, W. H. Ryu, J. L. Lee, W. I. Cho and B. W. Cho, J. Power Sources, 2006, 163, 185–190 CrossRef CAS.
- Y. Xiao, P. F. Wang, Y. X. Yin, Y. F. Zhu, X. N. Yang, X. D. Zhang, Y. S. Wang, X. D. Guo, B. H. Zhong and Y. G. Guo, Adv. Energy Mater., 2018, 8, 1800492 CrossRef.
- W. B. Hua, X. D. Guo, Z. Zheng, Y. J. Wang, B. H. Zhong, B. Z. Fang, J. Z. Wang, S. L. Chou and H. Liu, J. Power Sources, 2015, 275, 200–206 CrossRef CAS.
- J. L. Shi, D. D. Xiao, X. D. Zhang, Y. X. Yin, Y. G. Guo, L. Gu and L. J. Wan, Nano Res., 2017, 10, 4201–4209 CrossRef CAS.
- J. L. Shi, J. N. Zhang, M. He, X. D. Zhang, Y. X. Yin, H. Li, Y. G. Guo, L. Gu and L. J. Wan, ACS Appl. Mater. Interfaces, 2016, 8, 20138–20146 CrossRef CAS PubMed.
- Y. Xiao, X. D. Zhang, Y. F. Zhu, P. F. Wang, Y. X. Yin, X. N. Yang, J. L. Shi, J. Liu, H. L. Li, X. D. Guo, B. H. Zhong and Y. G. Guo, Adv. Sci., 2019, 6, 1801908 CrossRef PubMed.
- J. L. Shi, R. Qi, X. D. Zhang, P. F. Wang, W. G. Fu, Y. X. Xu, J. Xu, L. J. Wan and Y. G. Guo, ACS Appl. Mater. Interfaces, 2017, 9, 42829–42835 CrossRef CAS PubMed.
- W. Hua, M. Chen, B. Schwarz, M. Knapp, M. Bruns, J. Barthel, X. Yang, F. Sigel, R. Azmi, A. Senyshyn, A. Missiul, L. Simonelli, M. Etter, S. Wang, X. Mu, A. Fiedler, J. R. Binder, X. Guo, S. Chou, B. Zhong, S. Indris and H. Ehrenberg, Adv. Energy Mater., 2019, 9, 1803094 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0cc00327a |
| This journal is © The Royal Society of Chemistry 2020 |