
Cross-β amyloid nanotubes for hydrolase–peroxidase cascade reactions†
Ayan
Chatterjee
,
Syed Pavel
Afrose
,
Sahnawaz
Ahmed
,
Akhil
Venugopal
and
Dibyendu
Das
 *
*
Department of Chemical Sciences and Centre for Advanced Functional Materials, Indian Institute of Science Education and Research (IISER) Kolkata, Mohanpur-741246, India. E-mail: dasd@iiserkol.ac.in
First published on 28th February 2020
Abstract
Herein, we report the catalytic potential of short peptide based cross-β amyloid nanotubes with surface exposed histidine capable of binding hemin and showing facile cascade reactions, playing the dual roles of hydrolases and peroxidases, two of the most important classes of enzymes in extant biology. The activity of these simple systems exceeded those of modern and larger proteins like cytochrome C and hemoglobin. Further, evidence suggested that these self-assembled nanotubes foreshadow the process of intermediate channeling, a feature seen in the case of advanced enzymes.
Millions of years ago, inside Darwin's warm pond rich in nutrients, the origins of chemical evolution demanded the presence of a diverse chemical inventory and chemical networks.1,2 Computer simulation studies of these networks have argued for reaction diffusion models and supported the notion of diffusion differences to channel reaction substrates.3 Reactions such as oxidation of fuels achieved through a sequence of cascade reactions must have helped in the emergence of more complex products from simple precursors, thus enriching the inventory further.1–3 These chemical networks could indeed become mutually connected, eventually leading to stochastic innovation, best exemplified in Darwinian evolution.2 Chemical gradients and diffusion differences of reaction substrates are exploited in extant biology to master the art of single pot reactions in metabolic pathways. Enzymatic cascade reactions are carried out using proximally aligned biocatalysts where non-covalent associations and spatial co-localizations allow the product of one enzyme to act as a substrate for the other in the complex.3 Hence, this intermediate can channel to the adjacent active sites, resulting in enhancement of rates. This process is known as substrate channeling, which is advantageous in the context of minimizing, if not completely removing the expenses of reaching equilibrium with the bulk.
We asked whether a minimal system based on short peptide based assemblies can show cascade catalysis and benefit from such channeling as observed in modern enzymes.4–8 Many research laboratories including ours have reported the remarkable binding capabilities of short amyloid-forming peptides for diverse guests.4–13 Amyloid peptides and their assembly under prebiotic conditions have been shown recently by Riek and coworkers.1e,h,i Amyloid assemblies have also been elegantly scored for enzyme like activities and the results argue the evolution of enzymes from short assembling folds.14,15 We propose that exposed arrays of active site residues of amyloid assemblies coupled with their binding capabilities to attract co-factors/prosthetic groups can help amyloid to perform catalytic roles of multiple enzymes. Further, binding surfaces enriched with catalytic sites might promote channeling of cascade intermediates among the proximal sites (Fig. 1).
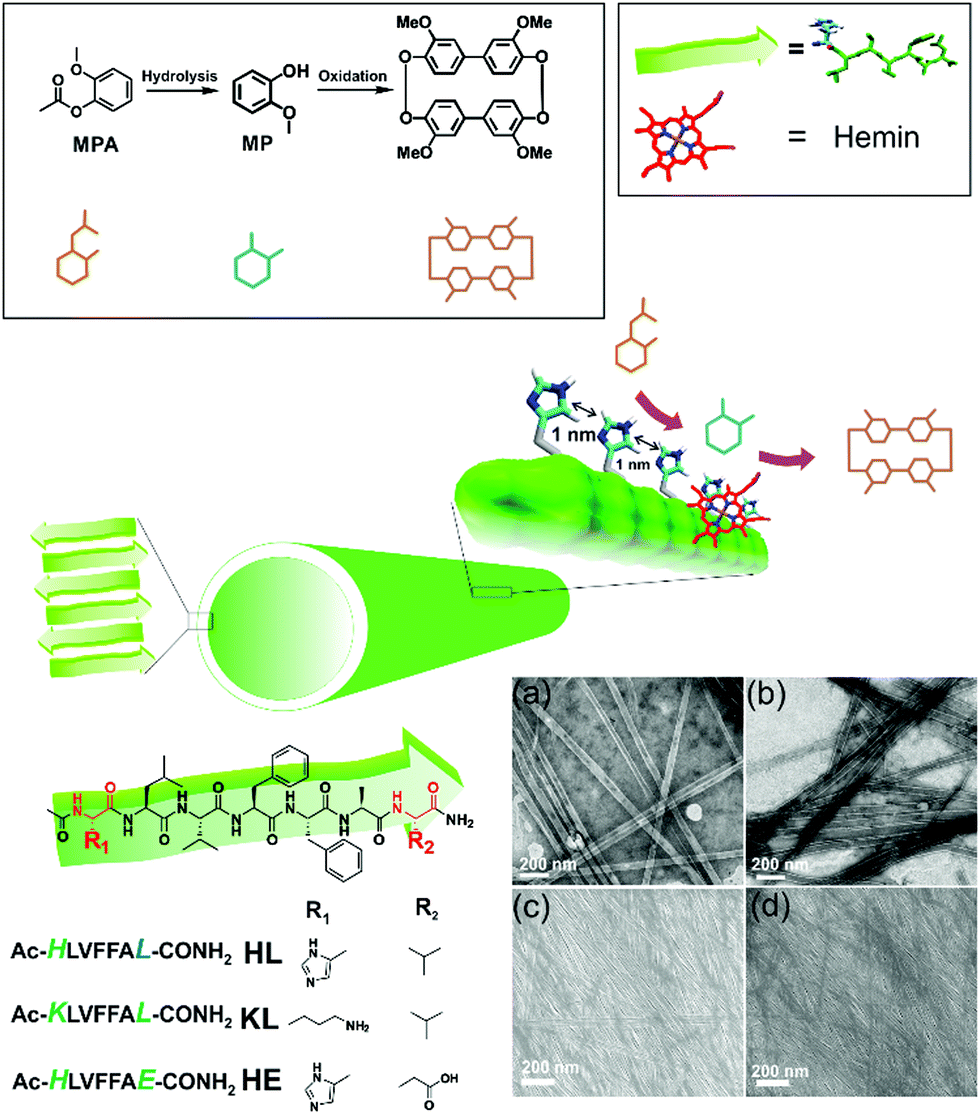 | ||
| Fig. 1 Scheme showing cascade reaction on nanotube surface, and chemical structures of amyloid, MPA, MP and the oxidation product. TEM and SEM images of self-assembled HL (a and c) and KL (b and d) respectively. | ||
We used a short peptide fragment (17LVFFA21) from the nucleating core of an Aβ (1–42) amyloid, an extensively studied sequence seen in the fibrillar deposits of Alzheimer's disease.4–12 This pentapeptide is known to access distinct morphologies with acute responsiveness to environmental switches.5–8,9a Ac-HLVFFAL-CONH2 (HL) was synthesized which assembles to form homogenous nanotubes with diameters of 35 ± 4 nm at pH 3 (Fig. 1a and c; Fig. S1–S3, ESI†). To investigate the exposure of histidines on their surfaces, the assemblies were incubated with negatively charged gold nanoparticles (Neg-AuNPs, Fig. 2 and Fig. S4, ESI;† for details of the pH effects on binding see Fig. S5A, ESI†). Homogenous tubular morphologies of similar dimensions with ordered arrays of AuNPs were observed under TEM and SEM (Fig. 2a–c, ESI†). A control experiment with cationic AuNPs did not yield any specific binding (Fig. S5B, ESI†), thus suggesting the presence of surface exposed histidines.
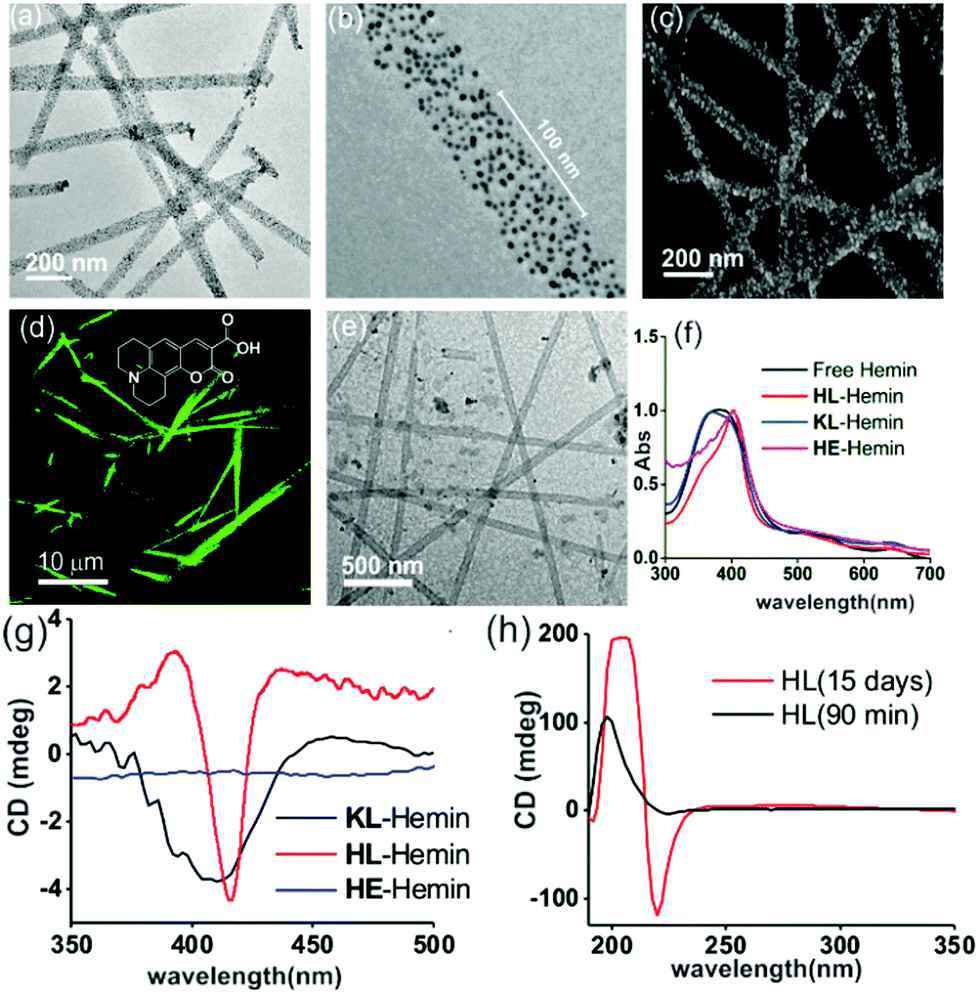 | ||
| Fig. 2 (a) TEM image of negative-AuNPs bound with HL and (b) an enlarged TEM image of a negative-AuNP bound with HL. (c) SEM image of AuNP–HL nanotubes. (d) CLSM image (λex = 488 nm) of Coumarin 343 dye bound to HL. (e) TEM image of hemin–HL. (f) Normalized UV-Vis spectra of free hemin, and hemin in the presence of HL, KL, and HE at pH 3. (g) CD spectra of hemin bound HL, KL, and HE. (h) CD spectra of HFIP treated HL after 90 min and 15 days. | ||
To probe the binding capability of these nanotubes, the hydrophobic fluorescent dye coumarin 343 was used as a model guest. Under a confocal fluorescence microscope (CLSM), fluorescent tubular structures were observed (Fig. 2d, controls in Fig. S6, ESI†). Blue shift in absorbance spectra was also observed upon binding (Fig. S7, ESI†). We expected that the nanotubes should also bind other small molecules like hemin – the prosthetic group of peroxidases and many metalloproteins. To create hemin bound assemblies, varying concentrations of hemin were mixed with 1 mM HL. The mole ratio varied in the range of ca. 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :150 of hemin:peptide with half of the peptide buried in the bilayer structure (Fig. S8A, ESI†).6 TEM showed no change in HL morphology after interaction with hemin (Fig. 2e). UV-Vis spectra show that unbound hemin had a Soret peak at 395 nm at pH 3 with a lower intensity peak at 365 nm, which is similar to the spectrum observed in water (Fig. S9, ESI†).16,17 These two major peaks suggested the presence of a dimer of hemin (μ-oxo bihemin) along with hemin-hydroxide (haematin) respectively. The 640 nm peak suggested the Q band value of the dimer.16a,17 In the presence of HL (pH 3), the Soret band of bound hemin significantly shifted to 404 nm with a shoulder of the hemin at 365 nm. The red shifted Soret peak suggested the formation of a hemin–histidine complex and lesser extent of hemin aggregation.16a,17 Circular dichroism (CD) showed an induced negative Cotton effect due to the achiral hemin, thus suggesting binding to the chiral surface of the nanotube (Fig. 2g). However, a crossover point at 404 nm suggested exciton coupling and hence indicated some dimerization.17b The peroxidase-like activity of these hemin–HL assemblies was then investigated. We used 2-methoxy phenol (MP, Fig. 1), a widely used substrate for monitoring peroxidase activity. Hemin–HL showed peroxidase like activity with kc = 14.5 ± 0.9 min−1 (Table S1; Fig. S10, ESI;† kinetic measurements were done at a fixed H2O2 concentration (30 mM); Fig. S11 (ESI†) for the effect of H2O2 concentration on the oxidation of MP).
:150 of hemin:peptide with half of the peptide buried in the bilayer structure (Fig. S8A, ESI†).6 TEM showed no change in HL morphology after interaction with hemin (Fig. 2e). UV-Vis spectra show that unbound hemin had a Soret peak at 395 nm at pH 3 with a lower intensity peak at 365 nm, which is similar to the spectrum observed in water (Fig. S9, ESI†).16,17 These two major peaks suggested the presence of a dimer of hemin (μ-oxo bihemin) along with hemin-hydroxide (haematin) respectively. The 640 nm peak suggested the Q band value of the dimer.16a,17 In the presence of HL (pH 3), the Soret band of bound hemin significantly shifted to 404 nm with a shoulder of the hemin at 365 nm. The red shifted Soret peak suggested the formation of a hemin–histidine complex and lesser extent of hemin aggregation.16a,17 Circular dichroism (CD) showed an induced negative Cotton effect due to the achiral hemin, thus suggesting binding to the chiral surface of the nanotube (Fig. 2g). However, a crossover point at 404 nm suggested exciton coupling and hence indicated some dimerization.17b The peroxidase-like activity of these hemin–HL assemblies was then investigated. We used 2-methoxy phenol (MP, Fig. 1), a widely used substrate for monitoring peroxidase activity. Hemin–HL showed peroxidase like activity with kc = 14.5 ± 0.9 min−1 (Table S1; Fig. S10, ESI;† kinetic measurements were done at a fixed H2O2 concentration (30 mM); Fig. S11 (ESI†) for the effect of H2O2 concentration on the oxidation of MP).
For tandem catalysis, the substrate undergoes consecutive reactions without the need for isolation of intermediates. Hence, MP was acetylated to form 2-methoxy phenyl acetate (MPA, Fig. 1) with the expectation that the hemin–HL nanotubes will first hydrolyze the ester to create the intermediate phenol MP which can be subsequently oxidized. The rate of oxidation will imply the rate of the tandem reaction. When hemin (0.01 mM) bound HL nanotubes were used for the acetylated phenol (MPA), rapid oxidation was observed (kc = 7.7 ± 0.7 min−1, Fig. 3a; see the ESI† for detailed kinetic treatment; Fig. S12 and Table S1; and see Fig. S13 (ESI†) for the effect of H2O2 concentration on the oxidation of MPA) at pH 3.18 From the pH rate profile, HL–hemin showed the highest activity at pH 3 (Fig. S11, ESI†). The reason for this could be attributed to the bundling of the HL nanotubes at higher pH as observed in TEM and scattering experiments (Fig. S14, ESI†). Varying hemin concentrations showed the highest activity at 0.01 mM hemin (Fig. S8B, ESI†). For controls, first Ac-KLVFFAL-CONH2 (KL, Fig. 1 and Fig. S15, ESI†) was synthesized.5–7,14aKL formed morphologically indistinguishable nanotubes (diameters of 36 ± 4 nm, Fig. 1b and d) and bound to Neg-AuNPs in a similar fashion to HL, suggesting the exposure of lysines on its nanosurface (Fig. S16, ESI†). Also, hemin binding was indicated by CD and UV-Vis spectra (Fig. 2f and g). Hemin bound lysine nanotubes showed low peroxidase activity with MP (kc = 0.5 ± 0.03 min−1 at pH 3, Table S1, ESI†) but substantially higher activity in the presence of histidine (1 mM, kc = 11.5 ± 0.8 min−1) as histidine is known to augment hemin activity (Table S1, ESI†).16a However, when the tandem reaction was monitored with acetylated phenol MPA as a substrate, hemin–KL nanotubes showed meagre activity of 0.08 ± 0.01 min−1 (Fig. 3 and Table S1, ESI†). Addition of 1 mM histidine to hemin–KL did not result in any notable increase of activity (kc = 0.21 ± 0.02 min−1, Table S1 and Fig. S17, ESI†). This significant decrease of ca. 37 fold in hemin–KL–histidine with respect to hemin–HL underpinned the importance of exposed histidines for tandem catalysis. Notably, despite the similar oxidation rates for MP by the hemin–HL and hemin–KL–histidine systems, the substantially lower rates for the tandem reaction suggest the importance of the hydrolysis step in the cascade process. Extant proteins like cytochrome C (CytC) and hemoglobin (Hb) showed high peroxidase activity with MP with kc values of 49.1 ± 5.3 min−1 and 12 ± 1.3 min−1. However, with MPA as a substrate, both proteins showed substantially decreased tandem rates of kc = 1.13 ± 0.09 min−1 and 0.84 ± 0.01 min−1, respectively, despite the presence of multiple histidines in their respective sequences. Remarkably, this hemin-short peptide-based system had 6.8 fold higher cascade activity than CytC, despite the oxidation rate being more than 3 times lower (Fig. 4a).
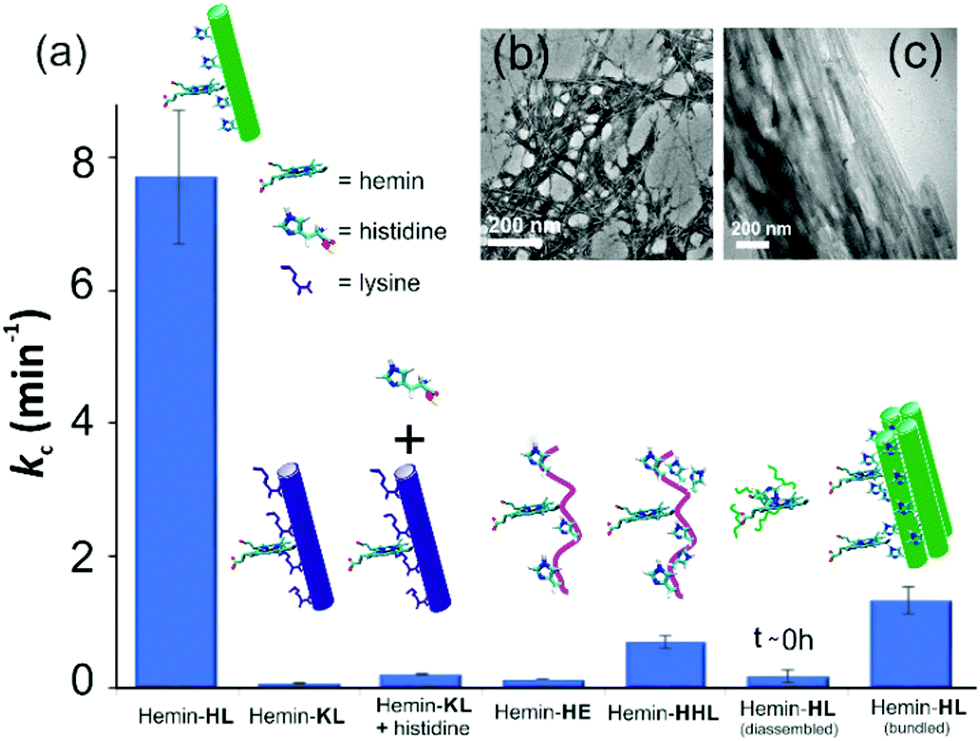 | ||
| Fig. 3 (a) Tandem activity with MPA. TEM images of (b) HE nanofibers and (c) bundled HL. [Hemin] = 0.01 mM, [peptide] = 1 mM, [H2O2] = 30 mM, [MPA] = 5 to 30 mM. Results are reported in triplicates. | ||
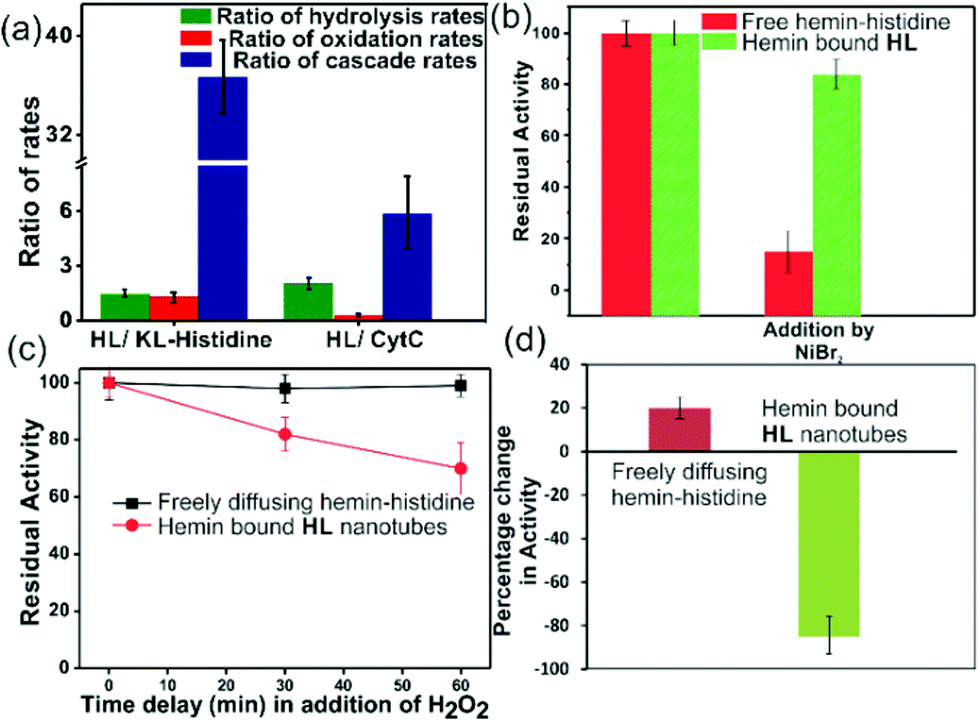 | ||
| Fig. 4 (a) Ratios of the rates of HL to KL–histidine systems and HL to CytC for hydrolysis, oxidation and cascade reactions. Residual activities of (b) free hemin–histidine and hemin–HL systems challenged by inhibitor NiBr2, (c) delayed addition of H2O2 to the free histidine hemin and hemin–HL systems, (d) percentage change in activity when MPA was added to HL or only histidine without hemin, and after 10 min delay, hemin was added. [Hemin] = 0.01 mM, [HL] = 1 mM, [histidine] = 1 mM, [H2O2] = 30 mM. | ||
To check the role of morphologies, we disassembled HL nanotubes with hexafluoroisopropanol (HFIP) as monitored from the loss of CD signals and TEM structures (Fig. 2h and Fig. S18, ESI†). With these disassembled HL nanotubes, hemin showed similar UV-Vis spectra to that observed for unbound hemin (Fig. S19, ESI†). The disassembled system showed 40 times lower activity (Fig. 3a). The terminal leucine of HL was mutated to form Ac-HLVFFAE-CONH2 (HE) which assembled to form nanofibers having diameters of 11 ± 2 nm (Fig. 3b and Fig. S20 and S21, ESI†). Hemin–HE showed a Soret peak at 404 nm, indicating histidine–hemin interaction, while CD did not show any induced peak (Fig. 2f and g). Hemin–HE showed decent peroxidase activity with MP (kc of 7.22 ± 0.65 min−1, Table S1, ESI†) but the tandem activity with MPA was substantially lower (kc = 0.14 ± 0.01 min−1, Fig. 3a and Table S1, ESI†). Addition of one more histidine residue at the N-terminus (Ac-HHLVFFAL-CONH2, HHL) resulted in nanofibrillar assembly (Fig. S22 and S23, ESI†) but still had 11 fold lower activity than HL (Fig. 3a and Table S1, ESI†). This result suggested the importance of a nanotubular morphology for cascade catalysis. To further investigate the role of nanosurfaces, Na2SO4 was added to induce bundling of nanotubes where the activity decreased to 1.32 ± 0.12 min−1 (Fig. 3c).5,6 Control experiments performed with free hemin–histidine systems in the presence of sodium sulfate did not show any decrease of activity, thus underpinning the role of the nanotubes for efficient cascade.
To probe the substantially faster cascade rates observed in the case of HL over the KL–histidine system (ca. 38 fold) and even enzymes such as CytC (ca. 6.8 fold), we compared the rates for hydrolysis reaction of MPA (Fig. 4a and Fig. S24–S26, ESI†). Interestingly, the ratios of the hydrolysis rates of HL to KL–histidine and CytC were found to be moderate (1.3 and 2 fold respectively). Hence, the reason for this significantly higher activation for cascade could be due to channeling in the histidine exposed HL nanotubes. To probe channeling, the cascade reaction was challenged by a reaction in the bulk environment.3 This method involved the addition of an inhibitor for the oxidation step (Fig. 4b).19 If intermediate channeling occurs, hemin–HL will show a lesser extent of inhibition compared to a reaction which happens in a freely diffusing bulk state. We used a combination of unbound hemin and free histidine as our freely diffusing model system which showed low but measurable rates with MPA (1.35 ± 0.11 min−1, Fig. S27, ESI†) only at higher pH 8, due to the nucleophilic hydrolytic and intrinsic peroxidase activities of histidine and hemin respectively.17d Ni2+ ions, a known inhibitor for the oxidation step of peroxidases, were added as a premixed solution with H2O2 to both the freely diffusing and hemin–HL systems.19 In the case of MP, the residual activity slumped down to almost 50% and 30% for the HL–hemin and freely diffusing systems, respectively (Fig. S28, ESI†), whereas for the cascade reaction with MPA, the activity of the freely diffusing system plummeted to 15% of the initial activity (Fig. 4b). However, for MPA with hemin–HL, the residual activity was still 85%, suggesting channeling in the nanotubes (Fig. 4b). Since H2O2 oxidizes MP, in a separate experiment we delayed H2O2 addition to allow the leakage of intermediate phenol, which is the substrate for hemin and hence would impede channeling. For the bulk, delayed addition of H2O2 did not have any effect on the tandem activity (Fig. 4c). Interestingly, for hemin–HL, the activity decreased significantly with time, suggesting the leakage of the cascade intermediate MP after hydrolysis (Fig. 4c). To investigate further, we induced the leakage of the intermediate by also delaying the association between hemin and HL. For this purpose, we added the substrate MPA to nanotubes without hemin, and after ca. 10 min delay for leaking, we added hemin. The cascade activity for this system decreased to 12 fold. However, a similar experiment with the freely diffusing system registered a slight increase in activity (Fig. 4d).
In summary, we have reported a short peptide-based amyloid with arrays of active site residues capable of binding prosthetic groups and displaying the catalytic potential of cascade reactions. With distances of ca. 1 nm between the residues,6 these peptide based short amyloid assemblies foreshadow the concept of intermediate channeling, which is observed in modern enzyme complexes. The system has some limitations in terms of pH. Current efforts are intended to develop systems with a greater substrate scope and higher pH tolerance.
DD is thankful to a Nanomission Grant (SR/NM/NS-1082/2015), DST, GOI, AC acknowledges CSIR, SPA acknowledges UGC and SA acknowledges NPDF (PDF/2017/000782), SERB.
Conflicts of interest
The authors declare no conflicts of interest.Notes and references
- (a) C. R. Woese, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 8742–8747 CrossRef PubMed; (b) J. Greenwald and R. Riek, J. Mol. Biol., 2012, 421, 417–426 CrossRef PubMed; (c) O. Carny and E. Gazit, FASEB J., 2005, 19, 1051–1055 CrossRef; (d) J. T. Goodwin, A. K. Mehta and D. G. Lynn, Acc. Chem. Res., 2012, 45, 2189–2199 CrossRef PubMed; (e) J. Greenwald, M. P. Friedmann and R. Riek, Angew. Chem., Int. Ed., 2016, 55, 11609–11613 CrossRef PubMed; (f) C. P. Maury, Origins Life Evol. Biospheres, 2009, 39, 141–150 CrossRef; (g) B. M. Rode, Peptides, 1999, 20, 773–786 CrossRef; (h) J. Greenwald, W. Kwiatkowski and R. Riek, J. Mol. Biol., 2018, 430, 3735–3750 CrossRef; (i) S. K. Rout, M. P. Friedmann, R. Riek and J. Greenwald, Nat. Commun., 2018, 9, 234 CrossRef PubMed; (j) J. E. Smith, A. K. Mowles, A. K. Mehta and D. G. Lynn, Life, 2014, 4, 887–902 CrossRef.
- J. A. Bradford and K. A. Dill, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 10098–10103 CrossRef.
- (a) I. Wheeldon, S. D. Minteer, S. Banta, S. C. Barton, P. Atanassov and M. Sigman, Nat. Chem., 2016, 8, 299–309 CrossRef; (b) Y. Gao, C. C. Roberts, J. Zhu, J.-L. Lin, C. A. Chang and I. Wheeldon, ACS Catal., 2014, 5, 2149–2153 CrossRef; (c) Y. Zhang, S. Tsitkov and H. Hess, Nat. Catal., 2018, 1, 276–281 CrossRef; (d) X. Zhao, H. Palacci, V. Yadav, M. M. Spiering, M. K. Gilson, P. J. Butler, H. Hess, S. J. Benkovic and A. Sen, Nat. Chem., 2018, 10, 311–317 CrossRef; (e) R. Guha, F. Mohajerani, M. Collins, S. Ghosh, A. Sen and D. Velegol, J. Am. Chem. Soc., 2017, 139, 15588–15591 CrossRef.
- G. Wei, Z. Su, N. P. Reynolds, P. Arosio, I. W. Hamley, E. Gazit and R. Mezzenga, Chem. Soc. Rev., 2017, 46, 4661–4708 RSC.
- N. Kapil, A. Singh and D. Das, Angew. Chem., Int. Ed., 2015, 54, 6492–6495 CrossRef.
- W. S. Childers, A. K. Mehta, R. Ni, J. V. Taylor and D. G. Lynn, Angew. Chem., Int. Ed., 2010, 49, 4104–4107 CrossRef.
- N. Kapil, A. Singh, M. Singh and D. Das, Angew. Chem., Int. Ed., 2016, 55, 7772–7776 CrossRef.
- S. Li, A. N. Sidorov, A. K. Mehta, A. J. Bisignano, D. Das, W. S. Childers, E. Schuler, Z. Jiang, T. M. Orlando, K. Berland and D. G. Lynn, Biochemistry, 2014, 53, 4225–4227 CrossRef.
- (a) A. Singh, N. Kapil, M. Yenuganti and D. Das, Chem. Commun., 2016, 52, 14043–14046 RSC; (b) B. G. Cousins, A. K. Das, R. Sharma, Y. Li, J. P. McNamara, I. H. Hillier, I. A. Kinloch and R. V. Ulijn, Small, 2009, 5, 587–590 CrossRef.
- (a) T. P. J. Knowles and R. Mezzenga, Adv. Matter, 2016, 28, 6546–6561 CrossRef; (b) A. J. Baldwin, T. P. J. Knowles, G. G. Tartaglia, A. W. Fitzpatrick, G. L. Devlin, S. L. Shammas, C. A. Waudby, M. F. Mossuto, S. Meehan, S. L. Gras, J. Christodoulou, S. J. Anthony-Cahill, P. D. Barker, M. Vendruscolo and C. M. Dobson, J. Am. Chem. Soc., 2011, 133, 14160–14163 CrossRef; (c) M. R. Elkins, T. Wang, M. Nick, H. Jo, T. Lemmin, S. B. Prusiner, W. F. DeGrado, J. Stöhr and M. Hong, J. Am. Chem. Soc., 2016, 138, 9840–9852 CrossRef CAS.
- (a) T. P. Knowles, A. W. Fitzpatrick, S. Meehan, H. R. Mott, M. Vendruscolo, C. M. Dobson and M. E. Welland, Science, 2007, 318, 1900–1903 CrossRef CAS; (b) J. Kaplanm and W. F. DeGrado, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 11566–11570 CrossRef; (c) C. B. Bell, J. R. Calhoun, E. Bobyr, P.-P. Wei, B. Hedman, K. O. Hodgson, W. F. DeGrado and E. I. Solomon, Biochemistry, 2009, 48, 59–73 CrossRef CAS PubMed.
- (a) C. M. Micklitsch, P. J. Knerr, M. C. Branco, R. Nagarkar, D. J. Pochan and J. P. Schneider, Angew. Chem., Int. Ed., 2011, 50, 1577–1579 CrossRef CAS; (b) M. P. Hendricks, K. Sato, L. C. Palmer and S. I. Stupp, Acc. Chem. Res., 2017, 50, 2440–2448 CrossRef CAS; (c) R. Freeman, M. Han, Z. Álvarez, J. A. Lewis, J. R. Wester, N. Stephanopoulos, M. T. McClendon, C. Lynsky, J. M. Godbe, H. Sangji, E. Luijten and S. I. Stupp, Science, 2018, 362, 808–813 CrossRef CAS.
- Y. Gao, F. Zhao, Q. Wang, Y. Zhang and B. Xu, Chem. Soc. Rev., 2010, 39, 3425–3433 RSC.
- (a) T. O. Omosun, M.-C. Hsieh, W. S. Childers, D. Das, A. K. Mehta, N. R. Anthony, T. Pan, M. A. Grover, K. M. Berland and D. G. Lynn, Nat. Chem., 2017, 9, 805–809 CrossRef CAS; (b) C. Zhang, R. Shafi, A. Lampel, D. MacPherson, C. Pappas, V. Narang, T. Wang, C. Madarelli and R. V. Ulijn, Angew. Chem., Int. Ed., 2017, 56, 14511–14515 CrossRef CAS; (c) C. M. Rufo, Y. S. Moroz, O. V. Moroz, J. Stöhr, T. A. Smith, X. Hu, W. F. DeGrado and I. V. Korendovych, Nat. Chem., 2014, 6, 303–309 CrossRef CAS PubMed; (d) C. Zhang, X. Xue, Q. Luo, Y. Li, K. Yang, X. Zhuang, Y. Jiang, J. Zhang, J. Liu, G. Zou and X. J. Liang, ACS Nano, 2014, 8, 11715–11723 CrossRef CAS.
- (a) M. O. Guler and S. I. Stupp, J. Am. Chem. Soc., 2007, 129, 12082–12083 CrossRef CAS PubMed; (b) N. Singh, M. Kumar, J. F. Miravet, R. V. Ulijn and B. Escuder, Chem. – Eur. J., 2017, 23, 981–993 CrossRef CAS PubMed; (c) J. Meeuwissen and J. N. H. Reek, Nat. Chem., 2010, 2, 615–621 CrossRef CAS.
- (a) Q. Wang, Z. Yang, X. Zhang, X. Xiao, C. K. Chang and B. Xu, Angew. Chem., Int. Ed., 2007, 46, 4285–4289 CrossRef CAS; (b) Q. G. Wang, Z. M. Yang, L. Wang, M. Ma and B. Xu, Chem. Commun., 2007, 1032–1034 RSC; (c) Q. Wang, L. Li and B. Xu, Chem. – Eur. J., 2009, 15, 3168–3172 CrossRef CAS.
- (a) P. K. Shantha, G. S. S. Saini, H. H. Thanga and A. L. Verma, J. Raman Spectrosc., 2001, 32, 159 CrossRef CAS; (b) S. Matile, N. Berova, K. Nakanishi, J. Fleischhauer and R. W. Woody, J. Am. Chem. Soc., 1996, 118, 5198–5206 CrossRef CAS; (c) G. J. Corban, S. K. Hadjikakou, A. C. Tsipis, M. Kubicki, T. Bakas and N. Had- jiliadis, New J. Chem., 2011, 35, 213–224 RSC; (d) T. Uno, A. Takeda and S. Shimabayashi, Inorg. Chem., 1995, 34, 1599–1607 CrossRef CAS.
- K. J. Laidler, Chemical Kinetics, Pearson Education, New Delhi, 1987 Search PubMed.
- F. Pintus, A. Mura, A. Bellelli, A. Arcovito, D. Spanò, A. Pintus, G. Floris and R. Medda, FASEB J., 2008, 275, 1201–1212 CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0cc00279h |
| This journal is © The Royal Society of Chemistry 2020 |