
Tin negative electrodes using an FSA-based ionic liquid electrolyte: improved performance of potassium secondary batteries†
Takayuki
Yamamoto  *a
and
Toshiyuki
Nohira
*a
*a
and
Toshiyuki
Nohira
*a
*a
and
Toshiyuki
Nohira
*a
Abstract
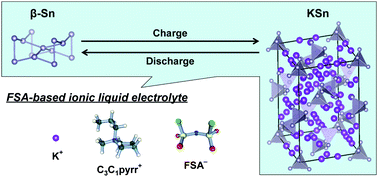
In this study, submicron-sized tin particles were used as the negative electrode material for potassium secondary batteries. With a bis(fluorosulfonyl)amide-based ionic liquid electrolyte, K[FSA]–[C3C1pyrr][FSA], the tin negative electrodes showed improved capacity retention of over 170 mA h (g-Sn)−1 after 100 cycles at room temperature.
 Please wait while we load your content...
Please wait while we load your content...

