
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Hydrophilic polyphosphoester-conjugated fluorinated chlorin as an entirely biodegradable nano-photosensitizer for reliable and efficient photodynamic therapy†
Zhiyong
Liu
a,
Mengsi
Wu
a,
Yudong
Xue
a,
Chao
Chen
 b,
Frederik R.
Wurm
*c,
Minbo
Lan
a and
Weian
Zhang
*a
b,
Frederik R.
Wurm
*c,
Minbo
Lan
a and
Weian
Zhang
*a
aShanghai Key Laboratory of Functional Materials Chemistry, East China University of Science and Technology, 130 Meilong Road, Shanghai 200237, China. E-mail: wazhang@ecust.edu.cn
bState Key Laboratory of Bioreactor Engineering Center, East China University of Science and Technology, 130 Meilong Road, Shanghai 200237, China
cMax Planck Institute for Polymer Research, Ackermannweg 10, Mainz 55128, Germany. E-mail: wurm@mpip-mainz.mpg.de
First published on 17th January 2020
Abstract
An entirely biodegradable nano-photosensitizer platform (PPE-FP2) was fabricated by conjugating the photosensitizer TFPC to hydrophilic polyphosphoesters (PPEs) for efficiently liberating photosensitizers at the tumor site. The complete biodegradability of PPE-FP2 avoided residual nanoparticles in vivo after therapy, realizing reliable and effective photodynamic therapy.
Nanocarriers such as liposomes,1,2 polymer micelles,3,4 MOFs5–7 and inorganic nanoparticles8,9 have been fabricated to deliver drugs to tumor cells via the enhanced permeability and retention (EPR) effect.10,11 In particular, pegylated nanoparticles have received massive attention. However, PEG cannot be biodegraded in vivo. Several concerns associated with the non-biodegradability of PEG have been raised, especially for long-term administration.12 The accumulation of PEG in vivo has been reported to result in intracellular vacuolation of organs such as the renal tubules, due to the stability of PEG.13 Furthermore, the formation of PEG antibodies has been observed, which can induce immune responses and accelerate the clearance of nanoparticles modified by PEG.14 To address the defects of non-biodegradable PEG, novel stealth polymers such as polyphosphoesters (PPEs),15 and also zwitterionic polymers,16–18 hydroxyethyl starch19,20 or dextrin21 have been proposed.
PPEs present great potential as a stealth material due to their biocompatibility and biodegradability.22 The backbone of PPEs is similar to biomacromolecules, such as DNA, RNA and teichoic acids and phosphorus is a fundamental element of bone making up 1% of the total human body mass.23,24 Therefore, the final degradation products of PPEs, phosphate or phosphonate, exist in or are well tolerated by the human body. Moreover, the backbone of PPEs can be cleaved under physiological conditions or recognized by enzymes, such as alkaline phosphatase, which is increased in tumor cells and can accelerate the hydrolysis of PPEs.25,26 Furthermore, the hydrolysis rate and functionality of PPEs can be controlled by side chain attachment resulting from reactive pendant groups of pentavalent phosphorus.27 Based on PPEs, Wooley et al. have designed several types of micelles used for drug delivery and gene therapy.28,29 In addition, Wang et al. also reported PPE-based nanoparticles for improving the antitumor effect of drugs.30,31 Importantly, as a stealth biomaterial, hydrophilic PPE has been verified to reduce protein adsorption and prevent nonspecific cellular uptake as well as PEG.32 Consequently, an effective and secure therapy could be achieved by a direct conjugation of PPE with hydrophobic agents.
Herein, we developed a fully biodegradable polymer photosensitizer by conjugating 7,8-dihydro-5,10,15,20-tetrakis(pentafluorophenyl)-21H,23H-porphine (TFPC) to the hydroxyl end groups of a telechelic poly(methyl ethylene phosphate) (PPE) as shown in Scheme 1. PPE is a hydrophilic polymer and after conjugation will self-assemble into larger aggregates with a PPE shell that should reduce protein adsorption. TFPC as a hydrophobic macrocyclic aromatic compound is easy to aggregate through π–π stacking and hydrophobic interaction; therefore, the polymer-photosensitizer conjugate can assemble into nanoparticles and accumulate in tumor sites by the EPR effect. Moreover, PPEs can be degraded in a physiological environment or by phosphatase, thus TFPC could be released from the nanoparticles for enhancing the sensitivity of photosensitizers to oxygen. Furthermore, the degradation of the nanoparticles can avoid their in vivo accumulation after therapy, reducing the side effects.
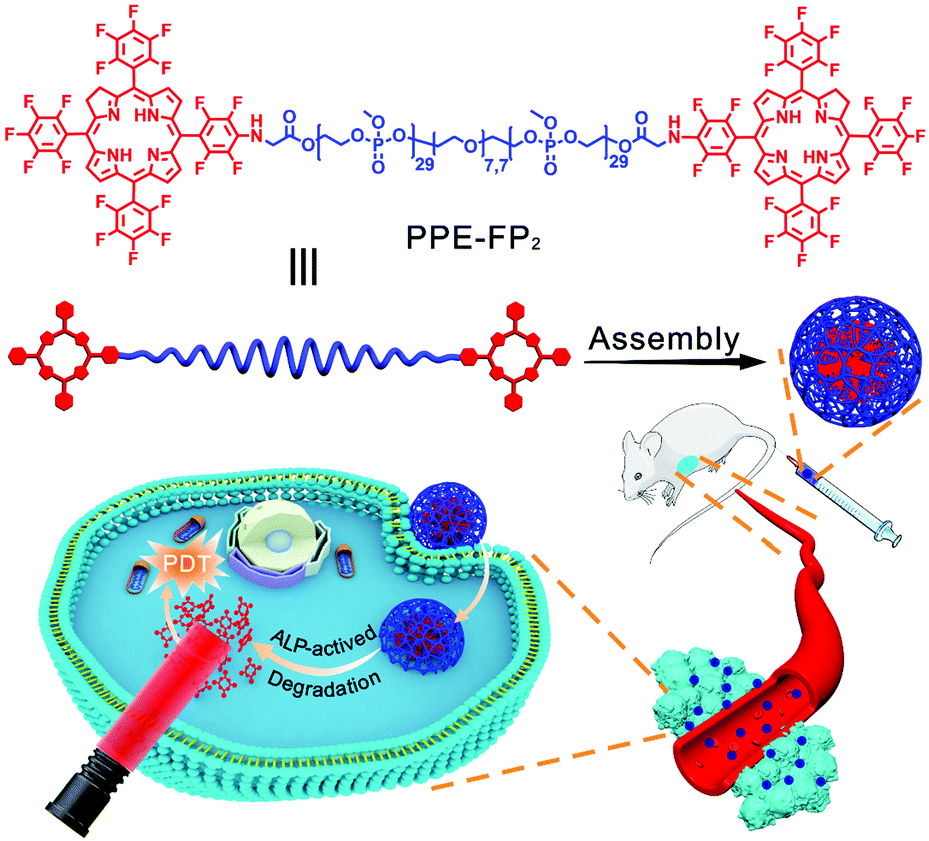 | ||
| Scheme 1 Schematic illustration of the self-assembly and application in photodynamic therapy of PPE-FP2. | ||
Meso-tetrakis(pentafluorophenyl)porphyrin (TFPP) was synthesized and then reduced by p-toluenesulfonylhydrazide to obtain the red-light-absorbing photosensitizer TFPC as shown in Scheme S1 (ESI†). Di-hydroxyl functionalized PPE with a molar mass of 4000 g mol−1 was synthesized by anionic ring-opening polymerization by an adapted literature protocol as the stealth material.33PPE-FP2 was prepared by conjugation of TFPC to PPE via esterification as shown in Scheme S1 (ESI†) and further assembled into spherical nanoparticles. Additionally, the PEG-analog, PEG-FP2 (Scheme S2, ESI†) was also prepared via a similar approach as a non-degradable control (detailed characterization is given in Fig. S1–S11, ESI†). Both polymer conjugates, PPE-FP2 and PEG-FP2 could be dispersed well in water and by the naked eye generated a stable dispersion with the typical yellowish color of the chlorin. In contrast, hydrophobic TFPC remained insoluble and sedimented. For both PPE-FP2 and PEG-FP2, spherical nanoparticles could be observed after the self-assembly by transmission electron microscopy (TEM) images (Fig. 1b and c). The size of the nanoparticles was relatively uniform for both PPE-FP2 and PEG-FP2 with average diameters of ca. 100 nm and 110 nm, respectively. Dynamic light scattering (DLS) further confirmed the size of the nanoparticles and supported the relatively narrow size distribution (Fig. 1d, an average hydrodynamic size of 160 nm, which is slightly larger than that determined by TEM, probably due to hydration). Subsequently, the potentials for photodynamic therapy and fluorescence imaging of PPE-FP2 were evaluated by UV absorbance spectrum in Fig. 1e and fluorescence emission spectroscopy in Fig. 1f, which show that PPE-FP2 nanoparticles have strong absorption (655 nm, ε(TFPC) = 1.62 × 104 L (mol−1 cm−1)) and emission (600–750 nm) in the far-red light region; therefore, PPE-FP2 nanoparticles could be used as a potential agent for the diagnosis of solid tumors.
 | ||
| Fig. 1 Characterization of PEG-FP2 and PPE-FP2 nanoparticles. (a) Photographs of TFPC, PEG-FP2 and PPE-FP2 in water. (b and c) TEM images of PEG-FP2 and PPE-FP2 nanoparticles. (d) Dynamic light scattering data of the nanoparticles. (e) UV spectra and (f) fluorescence spectra (ex: 425 nm) in ultrapure water. | ||
The in vitro biodegradability of the PPE-FP2 conjugate was separately assessed after direct dispersion in phosphate-buffered saline at pH = 7.4 and after the addition of alkaline phosphatase (ALP) while being shaken on a horizontal shaker at 37 °C. It is known that the concentration of ALP is increased in tumor cells, which should lead to an accelerated degradation of PPE.25 The degradation of PPE-FP2 in PBS with or without ALP was proven by UV absorbance and DLS measurements after certain time intervals (Fig. 2a–e and Fig. S12 and S13, ESI†). Compared with dispersion in PBS without ALP, the degradation rate of PPE-FP2 nanoparticles increased significantly in the presence of ALP, resulting in greater absorption reduction during 24 h as shown in Fig. 2b and c, which would promote photosensitizers to be effectively released from the PPE-FP2 nanoparticles at tumor sites. Degradation was followed by DLS (as shown in Fig. 2d and e and Fig. S13, ESI†): the size distribution of PEG-FP2 nanoparticles remained nearly unchanged, as they remained hydrolytically stable under these conditions, while the colloidal stability of the degradation products from PPE-FP2 decreased as the PPE hydrolyzed, resulting in aggregation of the hydrophobic chlorin. In the presence of ALP, PPE-FP2 nanoparticles were quickly destabilized during the first day, which further proved the biodegradation behavior of PPE accelerated by ALP. TEM images shown in Fig. S14–S18 (ESI†) also demonstrated that PPE-FP2 nanoparticles without the presence of ALP could maintain their distribution during the first day and degrade to irregular assembled aggregates after 3 days.
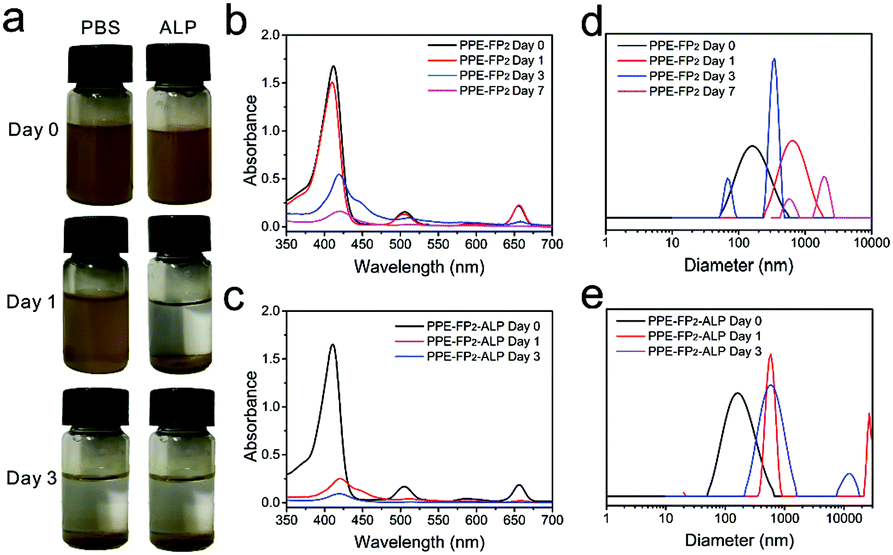 | ||
| Fig. 2 Stability evaluation of PPE-FP2 under different conditions. (a) Photos taken from the dispersions over time. UV spectrum of PPE-FP2 dispersed in PBS (b) and PBS with ALP (c). (d and e) Size distribution determined by DLS. | ||
Reactive oxygen species (ROS) generation of the nanoparticles was confirmed by using 1,3-diphenylisobenzofuran (DPBF) as an ROS indicator before investigating the in vitro and in vivo PDT performance.34 ROS generated by nanoparticles under 655 nm laser irradiation were recorded by a decrease of characteristic absorption of DPBF at ca. 415 nm (Fig. S19, ESI†). This result proved that both PPE-FP2 and PEG-FP2 nanoparticles could be used to as antitumor agents in PDT.
Confocal laser scanning microscopy (CLSM) was used to determine the cellular uptake of the nanoparticles. As shown in Fig. 3a, both PPE-FP2 and PEG-FP2 nanoparticles could be taken up effectively after incubating with 4T1 cells for 24 h. Interestingly, the fluorescence intensity of the cells treated with PPE-FP2 after 24 h was higher than that of the cells incubated with PEG-FP2, which might be a result of the degradation of PPE-FP2 after internalization into the tumor cells. Flow cytometry was used to study the cellular uptake of the chlorin conjugates (Fig. S20, ESI†). Gradually enhanced intracellular fluorescence intensity was observed with prolonged incubation times for both PPE-FP2 and PEG-FP2, indicating efficient internalization of the nanoparticles. Specifically, we found that the curves of fluorescence intensity of PPE-FP2 had two peaks at 4 h while there was only one peak at 24 h, indicating that PPE could be gradually degraded. Moreover, the higher fluorescence intensity of TFPC was observed in tumor cells treated with PPE-FP2 compared with cells incubated with PEG-FP2. These results suggested that PPE-FP2 could be gradually degraded to release TFPC in cells, thus reducing the aggregation induced quenching effect, which might enhance the PDT effect.
 | ||
| Fig. 3 Intracellular distribution and in vitro photodynamic therapy. CLSM images of (a) endocytosis by 4T1 cells and (b) intracellular ROS detected by DCFH-DA as a fluorescent probe. Cell viability against 4T1 cells (c) without and (d) with 655 nm laser (100 mW cm−2, 10 min) evaluated by MTT assay after incubation with PEG-FP2 and PPE-FP2 for 24 h. | ||
To gain insight into the PDT ability of PPE-FP2, the intracellular ROS generation of PPE-FP2 was evaluated by using 2,7-dichlorofluorescein diacetate (DCFH-DA) as an ROS probe.35 As shown in Fig. 3b, when combined with laser irradiation, the green fluorescence intensity of PPE-FP2 was significantly increased compared to that of the control, while PEG-FP2 exhibited a moderate enhancement of fluorescence. This result indicated the mass production of intracellular ROS induced by the nanoparticles under 655 nm laser irradiation, especially for biodegradable PPE-FP2. Based on these results, the in vitro PDT effect against 4T1 cells was verified via MTT assay. The cell viability approved in Fig. 3c demonstrated that PPE-FP2 and PEG-FP2 both have no significant cytotoxicity without laser irradiation on account of their good biocompatibility. However, when the cells incubated with nanoparticles were irradiated with a 655 nm laser, the cell viability showed a prominent decrease with the increase of the concentration of nanoparticles. Moreover, PPE-FP2 (IC50: 24 μg mL−1, calculated for chlorin concentration) exhibited a more efficient therapeutic effect than PEG-FP2 (IC50: 42 μg mL−1), which may have resulted from the biodegradation of PPE-FP2 accompanied by the liberation of TFPC.
Near infrared fluorescence agent Cyanine 7 (Cy7), a fluorescence quenching agent of TFPC based on fluorescence resonance energy transfer (FRET),36 was co-assembled with PPE-FP2 for in vivo fluorescence imaging as shown in Fig. 4a. The successful encapsulation of Cy7 is determined by UV spectra and DLS (Fig. S21 and S22, ESI†). The fluorescence imaging monitored in real-time revealed that nanoparticles could gradually accumulate into the tumor site with the extension of time and show a strong fluorescence signal at the tumor site at 24 h post-injection (p.i.). This consequence demonstrated that nanoparticles could effectively deliver TFPC into the tumor site through the EPR effect.
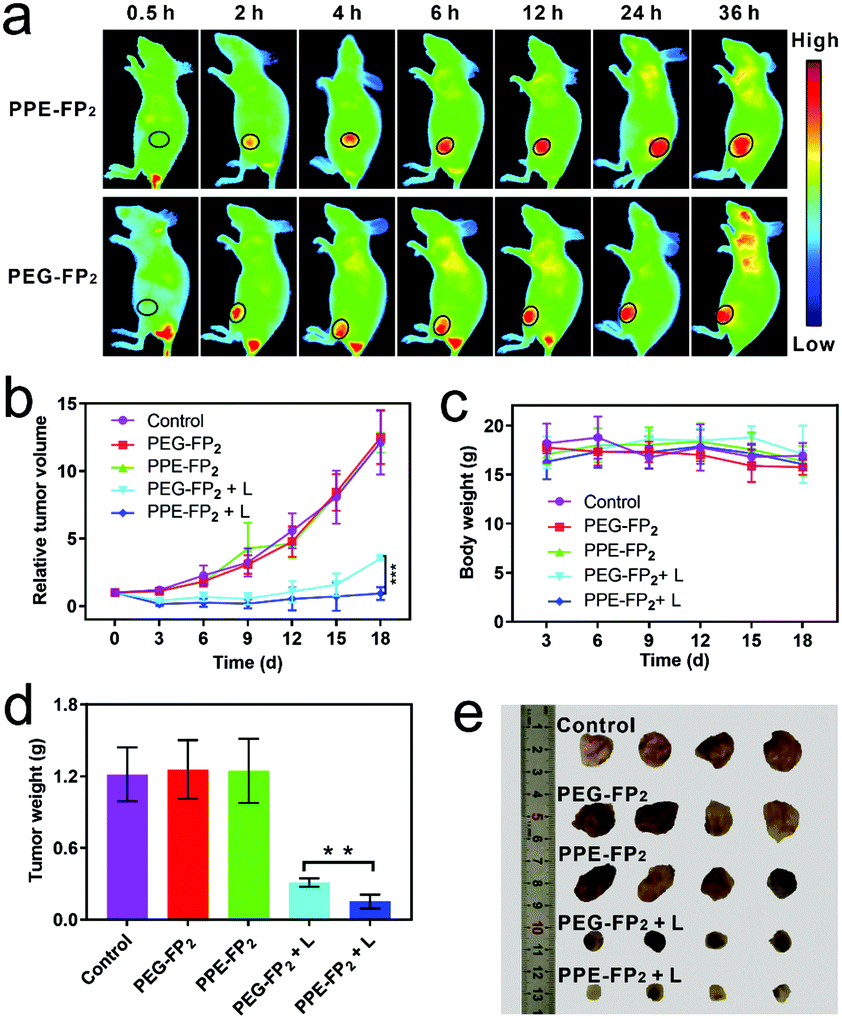 | ||
| Fig. 4 In vivo fluorescence imaging and anti-tumor efficacy against 4T1 tumors after intravenous injection of PBS (control), PEG-FP2 and PPE-FP2 nanoparticles at a TFPC-equivalent dose of 1 mg kg−1. (a) Cy7 (ex: 700 nm, em: 780 nm). (b) Relative tumor volumes. (c) Body weight and (d) tumor weight and (e) photographs of tumors. (n = 4, mean ± s.d., **P < 0.01, ***P < 0.001). | ||
Encouraged by the remarkable previous consequences, 4T1-tumor-bearing mice were used to investigate the in vivo anti-tumor performance. The mice were randomly divided into five groups: control, PEG-FP2, PPE-FP2, PEG-FP2 + L (655 nm laser irradiation), and PPE-FP2 + L. Relative tumor volume was recorded to study the suppressive effect as shown in Fig. 4b. The PEG-FP2 + L and PPE-FP2 + L groups showed a remarkable delay in tumor growth or tumor regression as compared with the control groups after 18 days. To further underline the efficiency of phototherapy, the tumors were excised from the mice and weighed to obtain the accurate tumor weight after the mice were sacrificed on day 18. As shown in Fig. 4d and e, tumors irradiated by a laser successfully reduced the size and weight, indicating the efficient photodynamic anti-tumor effect of PPE-FP2 and PEG-FP2. Besides, we found that there is a slow growth trend for tumors in the PEG-FP2 + L group while no obvious change in the PPE-FP2 + L group was observed, which could be attributed to the biodegradation behavior of PPE-FP2 with the enhanced PDT efficacy. Qualitative histological examinations were also carried out by hematoxylin and eosin (H&E) staining of tumor slices (Fig. S23, ESI†). Significant necrosis was observed on groups irradiated with a laser, especially for the PPE-FP2 + L group, proving the evident destruction of tumor cells by phototherapy. Fig. 4c and Fig. S24 (ESI†) revealed the minimal side effects of PEG-FP2 and PPE-FP2 during treatment.
A major benefit of PPE is the hydrolytic lability; thus, we increased the dose of the chlorin conjugates. The mice were sacrificed after two months, and then the kidneys and spleens were harvested and stained by H&E for toxicity analysis. Fig. S25 (ESI†) revealed that the kidneys, and spleens of mice administered with PEG-FP2 were damaged distinctly. In contrast, those treated with PPE-FP2 showed almost no damage. These results could be attributed to the degradation of PPEs avoiding the long-term accumulation of nanoparticles in the organs, indicating that the biodegradability of PPE endowed PPE-FP2 with excellent biological safety.
In summary, by conjugating TFPC to PPE, we developed a biodegradable nano-photosensitizer (PPE-FP2) for safe and effective photodynamic therapy. PPE-FP2 can be assembled into spherical nanoparticles and accumulate in tumor cells via the EPR effect. In addition, benefiting from the effective far-red absorption, PPE-FP2 could achieve deep tumor penetration. Importantly, as PPE-FP2 could be degraded, TFPC can be released from the assembly after cellular uptake, increasing the photodynamic efficacy of the PPE-conjugates, compared to the non-degradable PEGylated analog. In addition, as PPE-FP2 can be degraded and did not show any cytotoxicity in the absence of light, long-term in vivo accumulation is prevented. We believe that the nano-photosensitizers based on PPE are a new platform to design safe and more efficient photodynamic therapies. Together with the potential chemical functionality of PPEs, further targeting ligands or variation in their biodegradation profile can be installed, which will be studied in future projects.
This work was financially supported by the National Natural Science Foundation of China (No. 21875063), and the Open Access funding provided by the Max Planck Society.
Conflicts of interest
There are no conflicts to declare.Notes and references
- J. F. Lovell, C. S. Jin, E. Huynh, H. Jin, C. Kim, J. L. Rubinstein, W. C. Chan, W. Cao, L. V. Wang and G. Zheng, Nat. Mater., 2011, 10, 324–332 CrossRef CAS.
- X. Cai, D. Mao, C. Wang, D. Kong, X. Cheng and B. Liu, Angew. Chem., Int. Ed., 2018, 57, 16396–16400 CrossRef CAS.
- H. Park, W. Park and K. Na, Biomaterials, 2014, 35, 7963–7969 CrossRef CAS PubMed.
- Y. Li, T. Y. Lin, Y. Luo, Q. Liu, W. Xiao, W. Guo, D. Lac, H. Zhang, C. Feng, S. Wachsmann-Hogiu, J. H. Walton, S. R. Cherry, D. J. Rowland, D. Kukis, C. Pan and K. S. Lam, Nat. Commun., 2014, 5, 4712 CrossRef CAS.
- G. Lan, K. Ni, S. S. Veroneau, X. Feng, G. T. Nash, T. Luo, Z. Xu and W. Lin, J. Am. Chem. Soc., 2019, 141, 4204–4208 CrossRef CAS.
- G. Lan, K. Ni, Z. Xu, S. S. Veroneau, Y. Song and W. Lin, J. Am. Chem. Soc., 2018, 140, 5670–5673 CrossRef CAS PubMed.
- K. Zhang, X. D. Meng, Y. Cao, Z. Yang, H. F. Dong, Y. D. Zhang, H. T. Lu, Z. J. Shi and X. J. Zhang, Adv. Funct. Mater., 2018, 28, 1804634 CrossRef.
- Q. L. Wei, Y. Chen, X. B. Ma, J. F. Ji, Y. Qiao, B. Zhou, F. Ma, D. S. Ling, H. Zhang, M. Tian, J. Tian and M. Zhou, Adv. Funct. Mater., 2018, 28, 1704634 CrossRef.
- J. Song, X. Yang, Z. Yang, L. Lin, Y. Liu, Z. Zhou, Z. Shen, G. Yu, Y. Dai, O. Jacobson, J. Munasinghe, B. Yung, G. J. Teng and X. Chen, ACS Nano, 2017, 11, 6102–6113 CrossRef CAS PubMed.
- J. A. Hubbell and A. Chilkoti, Science, 2012, 337, 303–305 CrossRef.
- G. Song, L. Cheng, Y. Chao, K. Yang and Z. Liu, Adv. Mater., 2017, 29, 1700996 CrossRef PubMed.
- N. J. Butcher, G. M. Mortimer and R. F. Minchin, Nat. Nanotechnol., 2016, 11, 310–311 CrossRef CAS PubMed.
- J. Xu, J. Bussiere, J. Yie, A. Sickmier, P. An, E. Belouski, S. Stanislaus and K. W. Walker, Bioconjugate Chem., 2013, 24, 915–925 CrossRef CAS.
- M. Liu, P. Johansen, F. Zabel, J. C. Leroux and M. A. Gauthier, Nat. Commun., 2014, 5, 5526 CrossRef CAS.
- L. Zhang, D. Shi, C. Shi, L. Dong, X. Li and M. Chen, Macromol. Biosci., 2017, 17, 1600522 CrossRef.
- J. Zhao, Z. Qin, J. Wu, L. Li, Q. Jin and J. Ji, Biomater. Sci., 2017, 6, 200–206 RSC.
- L. Li, Y. Song, J. L. He, M. Z. Zhang, J. Liu and P. H. Ni, J. Mater. Chem. B, 2019, 7, 786–795 RSC.
- S. Iwasaki, H. Kawasaki and Y. Iwasaki, Langmuir, 2019, 35, 1749–1755 CrossRef CAS PubMed.
- Y. Tang, Y. Li, S. Li, H. Hu, Y. Wu, C. Xiao, Z. Chu, Z. Li and X. Yang, Nanoscale, 2019, 11, 6217–6227 RSC.
- K. Zhao, D. Li, W. Xu, J. Ding, W. Jiang, M. Li, C. Wang and X. Chen, Biomaterials, 2017, 116, 82–94 CrossRef CAS PubMed.
- H. P. Li, Z. W. Zhou, F. R. Zhang, Y. X. Guo, X. Yang, H. L. Jiang, F. Tan, D. Oupicky and M. J. Sun, Nano Res., 2018, 11, 4627–4642 CrossRef CAS.
- H. Elzeny, F. Zhang, E. N. Ali, H. A. Fathi, S. Zhang, R. Li, M. A. El-Mokhtar, M. A. Hamad, K. L. Wooley and M. Elsabahy, Drug Des., Dev. Ther., 2017, 11, 483–496 CrossRef CAS.
- K. Hirota, T. Hristova, V. Mitova, T. Koda, M. Fushimi, M. Kuniya, K. Makino, H. Terada, R. Cherkezova, S. Yusa, N. Koseva and K. Troev, Anticancer Res., 2016, 36, 1613–1620 CAS.
- R. Moriyama, Y. Iwasaki and D. Miyoshi, J. Phys. Chem. B, 2015, 119, 11969–11977 CrossRef CAS PubMed.
- X. Liu, Y. Li, X. Tan, R. Rao, Y. Ren, L. Liu, X. Yang and W. Liu, Biomaterials, 2018, 157, 136–148 CrossRef CAS.
- M. Yao, Y. Ma, H. Liu, M. I. Khan, S. Shen, S. Li, Y. Zhao, Y. Liu, G. Zhang, X. Li, F. Zhong, W. Jiang and Y. Wang, Biomacromolecules, 2018, 19, 1130–1141 CrossRef CAS PubMed.
- K. N. Bauer, L. Liu, D. Andrienko, M. Wagner, E. K. Macdonald, M. P. Shaver and F. R. Wurm, Macromolecules, 2018, 51, 1272–1279 CrossRef CAS.
- Y. P. Borguet, S. Khan, A. Noel, S. P. Gunsten, S. L. Brody, M. Elsabahy and K. L. Wooley, Biomacromolecules, 2018, 19, 1212–1222 CrossRef CAS.
- F. Zhang, J. A. Smolen, S. Zhang, R. Li, P. N. Shah, S. Cho, H. Wang, J. E. Raymond, C. L. Cannon and K. L. Wooley, Nanoscale, 2015, 7, 2265–2270 RSC.
- P. Pei, C. Sun, W. Tao, J. Li, X. Yang and J. Wang, Biomaterials, 2019, 188, 74–82 CrossRef CAS.
- H. Jin, M. Sun, L. Shi, X. Zhu, W. Huang and D. Yan, Biomater. Sci., 2018, 6, 1403–1413 RSC.
- S. Schottler, G. Becker, S. Winzen, T. Steinbach, K. Mohr, K. Landfester, V. Mailander and F. R. Wurm, Nat. Nanotechnol., 2016, 11, 372–377 CrossRef.
- C. Bernhard, K. N. Bauer, M. Bonn, F. R. Wurm and G. Gonella, ACS Appl. Mater. Interfaces, 2019, 11, 1624–1629 CrossRef CAS PubMed.
- L. Huang, Z. Li, Y. Zhao, J. Yang, Y. Yang, A. I. Pendharkar, Y. Zhang, S. Kelmar, L. Chen, W. Wu, J. Zhao and G. Han, Adv. Mater., 2017, 29, 1604789 CrossRef PubMed.
- M. Huo, L. Wang, Y. Chen and J. Shi, Nat. Commun., 2017, 8, 357 CrossRef PubMed.
- G. X. Liu, Y. M. Zhang, X. F. Xu, L. Zhang, Q. L. Yu, Q. Zhao, C. Y. Zhao and Y. Liu, Adv. Opt. Mater., 2017, 5, 1700770 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0cc00142b |
| This journal is © The Royal Society of Chemistry 2020 |