
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA simplicity-guided cocktail approach toward multicolor fluorescent systems†
Gaowa
Naren
,
Shiming
Li
and
Joakim
Andréasson
 *
*
Department of Chemistry and Chemical Engineering, Chemistry and Biochemistry, Chalmers University of Technology, SE-41296 Göteborg, Sweden. E-mail: a-son@chalmers.se
First published on 24th February 2020
Abstract
A molecular cocktail containing two photochromic diarylethene derivatives that displays multicolor emission spanning blue-green to orange in a color-correlated fashion has been devised. The function does not rely on excited state communication such as energy transfer reactions, which is the typical case for similar systems. Instead, harnessing the intrinsic fluorescent and photochromic properties of the two individual diarylethene derivatives run in parallel is enough to realize the color changes. This offers an extremely flexible situation as for the choice of the fluorophores and their respective concentrations. The function is conveniently demonstrated in bulk solution at μM concentrations, where a single light source serves as the color changing stimulus.
Over the last decade, there has been an increasing interest in molecular systems displaying stimuli-responsive changes in the emission color. Main drivers for this development are potential applications in optical data storage, bioimaging, super-resolution microscopy, sensors, anticounterfeiting, and more.1–11 Compared to binary on–off (single-color) fluorescence switching, stimuli-responsive multicolor systems offer many upsides such as allowing for ratiometric methods,12–15 full-color reproduction,16–18 and the possibility to introduce a high degree of unpredictability in the associated spectral changes, a highly sought-after property in data-security contexts.1,19,20
For multicolor fluorescence photoswitching, the color changes can be brought about in two principally different ways: color-specific and color-correlated switching.4 The latter allows for a much more distinct color change, as the increase in emission in one wavelength region is concerted, or correlated, with the disappearance of the emission in another wavelength region. A common strategy for color-correlated systems is to make use of FRET processes,21–24 typically by employing a “static” fluorescent dye as the energy donor and a molecular photoswitch (photochromic molecule) as the corresponding acceptor. Isomerization of the photochromic acceptor between the colorless passive form and the colored, fluorescent FRET-active form allows in principle for convenient toggling of the emission between 100% donor-centered to 100% acceptor-centered. Efficient FRET reactions, however, require the donor and the acceptor in close proximity, typically at around 40 Å or closer. Several supramolecular and covalently linked constructs have been presented,16,25–31 the common denominator being a careful organization of the system into a suitable geometry that allows for efficient FRET reactions to occur. Most often these approaches imply complicated synthetic procedures and the loss of system flexibility. Here we report a novel, extremely facile means of color-correlated photoswitching by simply mixing a cocktail of two photochromic diarylethene (DAE) derivatives32 in acetonitrile bulk solution at μM concentration. We harness merely the intrinsic fluorescent and photochromic properties of the two photoswitches without the need to fine-tune excited state reactions such as the abovementioned FRET processes. In addition, the system is controlled by a single light source at 381 nm triggering the color change, and a second light source at 410 nm for emission readout with virtually non-destructive character.
The structures and the isomerization scheme of the two DAE derivatives used in this study are shown in Scheme 1. Herein, we refer to the compounds as Dasy (DAE asymmetric) and DAE9 (adopted from the nomenclature in the original report).33 Both exist in an open isomeric form absorbing almost exclusively in the UV region (see Fig. 1 for spectral properties). UV exposure triggers isomerization from the open to the closed form, and the reverse reaction is induced by visible light. Dasy is fluorescent in the open isomeric form Dasy(o) with a quantum yield of 0.11 whereas the closed ditto Dasy(c) displays no detectable emission. The opposite is true for DAE9 where there is no detectable emission from DAE9(o) but DAE9(c) emits fluorescence with a quantum yield of 0.37. This value is significantly lower compared to the fluorescence quantum yield in 1,4-dioxane (0.61).33 Similar DAE derivatives have been reported to show the same trend, that is, a decrease in the fluorescence quantum yield with increasing solvent polarity.34
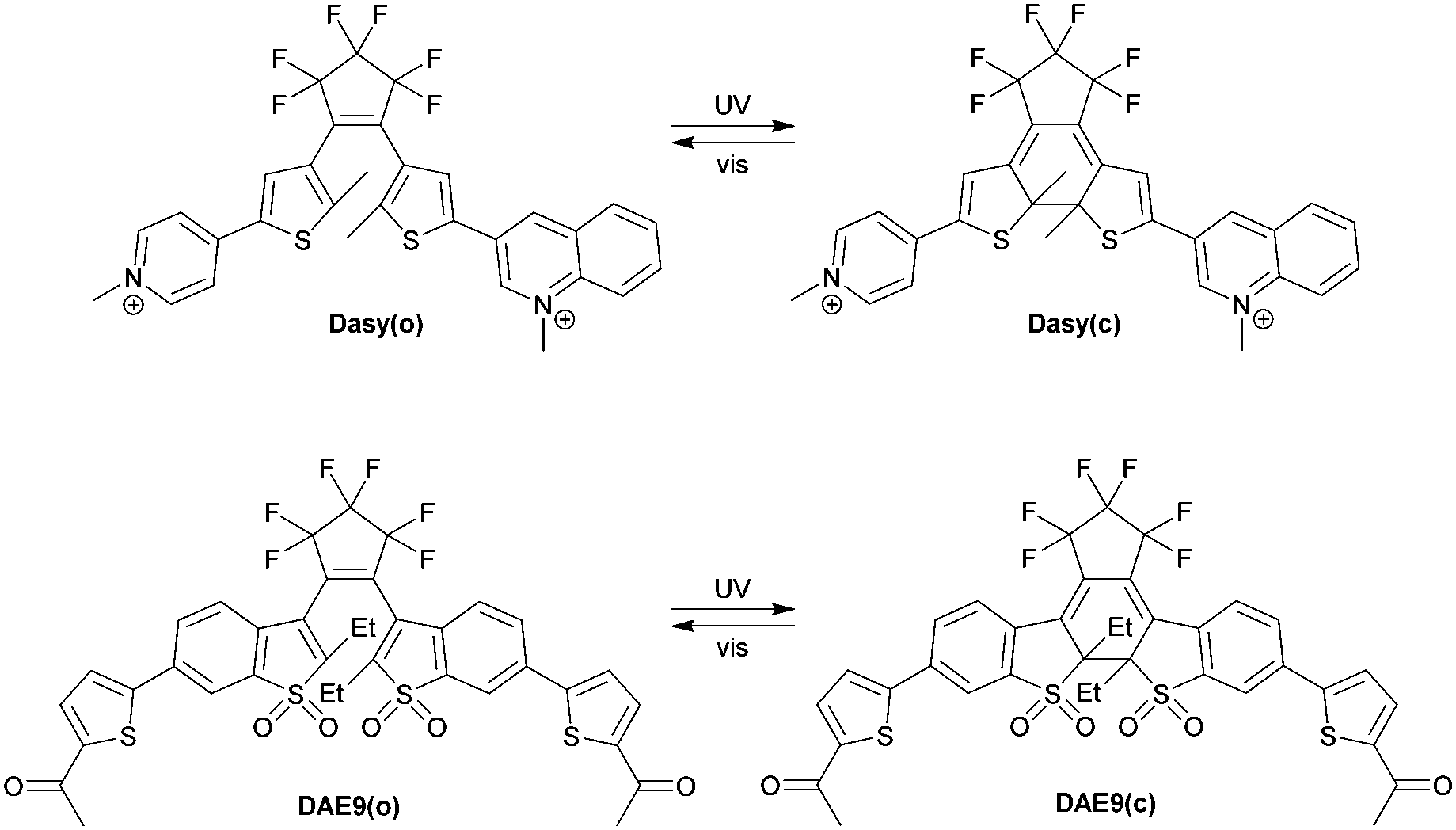 | ||
| Scheme 1 Structures and isomerization scheme of the studied compounds. | ||
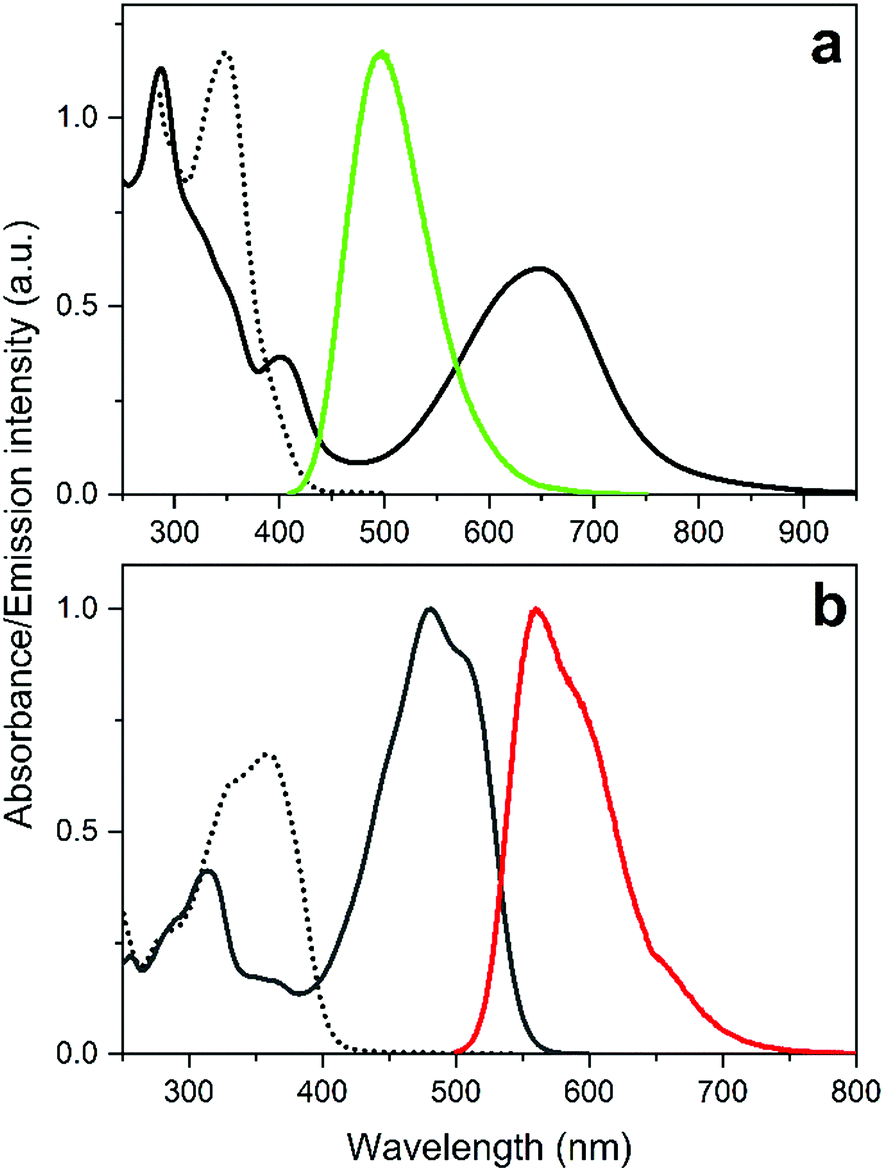 | ||
| Fig. 1 Absorption and emission spectra of Dasy (a) and DAE9 (b). Absorption spectra of the open isomers (dotted black lines) and the closed isomers (solid black lines) are shown together with the emission spectra (green solid line for Dasy(o) and red solid line for DAE9(c)). | ||
As for the emission colors, Dasy(o) emits blue-green light centered at around 497 nm, and DAE9(c) emits in the orange region with emission maximum at 561 nm. The CIE coordinates of the respective spectra are shown in Fig. S12, ESI.† We would like to emphasize that a fluorescence quantum yield of 0.11 is unusually high for the open isomer of a regular perfluoro DAE derivative. In fact, we know of only two reported examples with a higher number.‡![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 35,36 These compounds, however, suffer from a UV-induced photostationary state poorly enriched in the closed form, implying that “on–off” switching of the emission is not possible.
35,36 These compounds, however, suffer from a UV-induced photostationary state poorly enriched in the closed form, implying that “on–off” switching of the emission is not possible.
Exposing a cocktail solution of Dasy(o) and DAE9(o) to UV light would imply initial emission of blue-green fluorescence from Dasy(o) only. This emission will gradually disappear, as UV exposure triggers also the isomerization to the non-fluorescent Dasy(c) isomer. Concomitant to the decrease in blue-green emission intensity, an increase in the orange emission from DAE9(c) is expected, due to the UV-induced formation of this species. Extended UV irradiation would eventually yield a total disappearance of the blue-green emission (given that the photostationary distribution of Dasy is 100% shifted to the non-fluorescent Dasy(c) isomer) and exclusively intense orange emission from DAE9(c). Please note that this color change is expected to occur without the need for FRET reactions. It is simply a matter of balancing the rates of the two isomerization reactions, Dasy(o) → Dasy(c) and DAE9(o) → DAE9(c) allowing for a continuous color change. The choice of irradiation wavelength indeed allows for tuning the relative rates of these isomerization reactions (Fig. S13, ESI† and text below).
As for the reading process, that is, excitation for emission readout, a constant overall emission intensity throughout the process is desirable. Moreover, reading the sample in a non-destructive fashion, implying that the excitation light for emission readout does not induce changes in the isomeric distribution, would also add to the appeal. The former matter reduces to choosing the excitation wavelength as to match the brightness of the two fluorophores, whereas the latter implies exciting the cocktail at a wavelength with minimal absorption of the photochromically active species. We have detailed in the ESI,† how the photophysical properties of the photoswitches are used as a guide to the experimental conditions for optimal performance according to above.
Fig. 2 shows the evolution of the overall emission spectra of a sample containing ca. 35 μM Dasy and 2.3 μM DAE9 with increasing exposure time to 381 nm UV light. Excitation for emission readout was done at 410 nm. It is clearly seen that the initial spectrum (green) contains no or only minor contribution from DAE9(c) but is instead dominated by Dasy(o), whereas the final spectrum (red) shows fluorescence from almost exclusively DAE9(c). Moreover, the overall emission intensities are comparable at all times as shown in Fig. S14, ESI,† and the color change occurs in a continuous fashion from blue-green to orange, as is evident from Fig. S15, ESI.† This clearly shows that the rates of the isomerization reactions Dasy(o) → Dasy(c) and DAE9(o) → DAE9(c) occur on a similar timescale, which is also seen by comparing the individual isomerization rates from UV-vis absorption measurements where the rates differ by only a factor 2 (see Fig. S13, ESI†). The isomerization quantum yields for the closing reactions are 0.52 and 0.13 for Dasy and DAE9, respectively. Resetting the sample to the initial state containing only Dasy(o) and DAE9(o) is conveniently done by exposure to visible light, in this case at 523 nm, allowing for repeated operation with excellent reproducibility (Fig. S16 and S17, ESI†). The sample can be exposed to at least five switching cycles without substantial photodegradation (Fig. S18, ESI†).
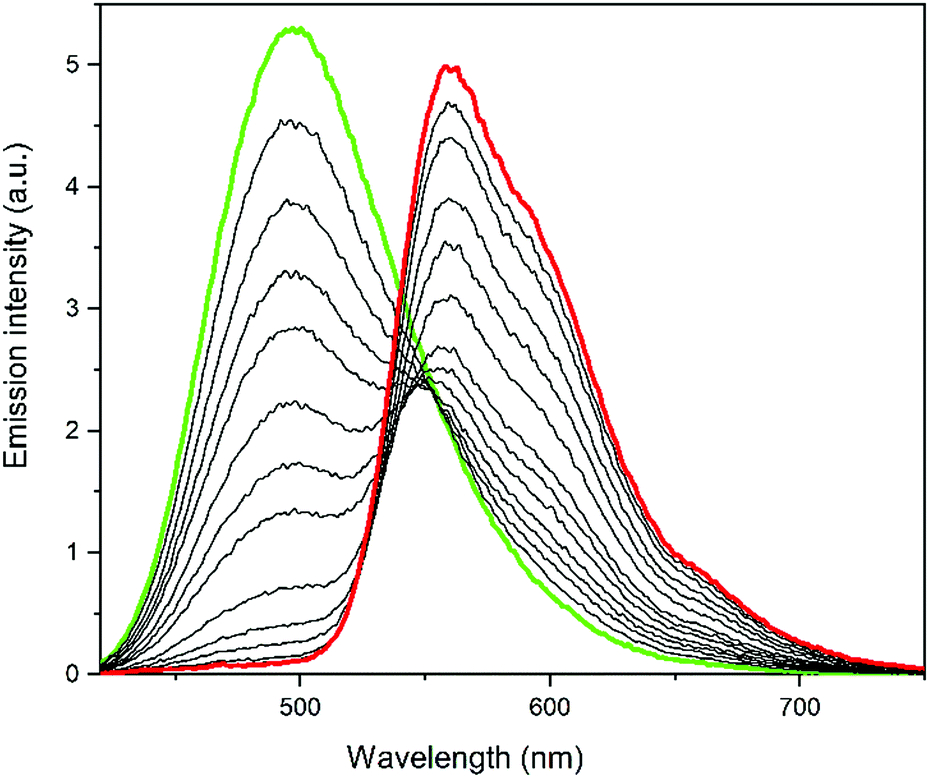 | ||
| Fig. 2 Time evolution of the emission spectra for the Dasy + DAE9 cocktail in acetonitrile under exposure to 381 nm UV light for isomerization purpose. All spectra were recorded upon excitation at 410 nm after exposure to 381 nm light for 0 s (green line), 5 s, 10 s, 15 s, 20 s, 30 s, 40 s, 50 s, 80 s, 110 s, 140 s, 200 s, 260 s, and 350 s (red line). | ||
The stability of the system with respect to color changes over time both in the absence and in the presence of the excitation light for emission readout was investigated (Fig. S19–S23, ESI†). The former reflects the thermal stability of the system when the photoswitches are in the closed isomeric form. Here, samples of DAE9(o) and Dasy(o) were converted to the photostationary state using UV light at 365 nm (both photoswitches display virtually quantitative conversion to the closed isomers) and left for 24 hours in the dark. No detectable changes in the absorption spectra were observed for DAE9, whereas Dasy experienced ca. 10% decrease in the absorption of the closed isomer. The absorbance in the wavelength region 330–380 nm where Dasy(o) displays its characteristic strong absorption band also decreased, strongly signalling that the spectral changes are due to the irreversible formation of a by-product.37 The system displays excellent color stability with respect to excitation for emission readout, that is, only minor changes in the emission spectra were observed after recording up to 80 spectra with excitation at 410 nm. There is, of course, an inevitable trade-off between absolute emission intensity and color stability, and in this study the excitation wavelength was chosen as to optimize the color stability without sacrificing too much in the signal-to-noise of the fluorescence signal.
In order to demonstrate that the observed color changes do not rely on FRET reactions of any kind, time-resolved single photon counting experiments were undertaken (see Fig. S24 and S25, ESI†). The fluorescent lifetimes of the monomers alone in acetonitrile are 1.8 ns for DAE9(c) and 5.4 ns for Dasy(o). The lifetimes for DAE9(c) in the cocktail together with Dasy(o) and Dasy(c) are both 1.8 ns, whereas Dasy(o) has lifetimes of 5.8 ns and 5.7 ns in the cocktail with DAE9(o) and DAE9(c), respectively. Thus, it is clear that quenching by FRET reactions do not occur.
Finally, the choice of photoswitches should be commented on. The first prerequisite is that one of the two must be emissive in the colorless form, whereas the other must be emissive in the colored form. Indeed, a photoswitch that displays intense emission in both isomeric forms would be usable too, but to date, there are no photochromic compounds with this favourable property. Moreover, it would imply a loss in the overall flexibility as the total concentration of the two emissive species would remain constant through the isomerization process. Using Dasy and DAE9 demonstrates the usefulness of the cocktail approach to finetune both the relative concentrations and the excitation wavelengths to compensate for differences in the emission- and the isomerization quantum yields of the ingredient photoswitches. Please note that although Dasy(o) used herein displays unusually high fluorescence quantum yield compared to the typical open form of any DAE derivative, an equivalent function would be achievable also for less fluorescent compounds, but then at a higher concentration to compensate. Alternatively, a covalently linked dyad comprising a fluorophore covalently linked to a non-fluorescent photochromic compound to constitute a photoswitchable FRET (or photoinduced electron transfer) donor–acceptor pair could be used. Note, however, that this approach would impose additional constraints to the design criteria, as the excited state interactions between the donor and the acceptor would have to be carefully tuned.
Moreover, the structural properties of Dasy and DAE9 differ, in particular with respect to Dasy being a dication whereas DAE9 is charge neutral. This presents challenges as for many approaches to self-assemble both compounds in confined volumes, which is a requirement for efficient FRET reactions to occur. The herein described approach overcomes also this limitation. It should be noted, however, that the ease of operation requires a medium in which the two photoswitches are fairly evenly distributed, as regions of high local concentration of both chromophores will interfere with the overall function by the onset of excited state reactions such as FRET and photoinduced electron transfer.
In summary, we have presented a novel approach to multicolor fluorescent photoswitching in a color-correlated fashion. The function does not rely on FRET reactions to occur, which allows for an extremely simple experimental protocol where two fluorescent DAE derivatives are mixed in bulk acetonitrile solution at μM concentrations. Hence, tedious synthesis of covalently linked constructs or the formation of supramolecular systems are avoided. A single light source is responsible for the isomerization-induced color changes and the emission readout process is virtually non-destructive. The all-photonic nature of the external stimulus presents the typical advantages (waste-free, non-invasive, and remote operation together with the unsurpassed spatiotemporal control) compared to systems that are driven by chemical or mechanical triggers.
JA acknowledges financial support from the Swedish Research Council VR (Grant # 2016-0360). The authors thank Dr Tamara Pace and Dr Vilhelm Müller for participating in the initial studies of Dasy.
Conflicts of interest
There are no conflicts to declare.Notes and references
- J. Andréasson and U. Pischel, Chem. Soc. Rev., 2018, 47, 2266–2279 RSC.
- N. Hildebrandt, C. M. Spillmann, W. R. Algar, T. Pons, M. H. Stewart, E. Oh, K. Susumu, S. A. Diaz, J. B. Delehanty and I. L. Medintz, Chem. Rev., 2017, 117, 536–711 CrossRef CAS PubMed.
- X. S. Hou, C. F. Ke, C. J. Bruns, P. R. McGonigal, R. B. Pettman and J. F. Stoddart, Nat. Commun., 2015, 6, 6884 CrossRef PubMed.
- D. Kim and S. Y. Park, Adv. Opt. Mater., 2018, 6, 1800678 CrossRef.
- C. H. Li and S. Y. Liu, Chem. Commun., 2012, 48, 3262–3278 RSC.
- V. K. Praveen, C. Ranjith and N. Armaroli, Angew. Chem., Int. Ed., 2014, 53, 365–368 CrossRef CAS PubMed.
- S. Silvi, M. Baroncini, M. La Rosa and A. Credi, Top. Curr. Chem., 2016, 374, 65 CrossRef PubMed.
- Z. Y. Tian, W. W. Wu and A. D. Q. Li, ChemPhysChem, 2009, 10, 2577–2591 CrossRef CAS PubMed.
- Y. L. Zhang, K. Q. Zhang, J. Wang, Z. Y. Tian and A. D. Q. Li, Nanoscale, 2015, 7, 19342–19357 RSC.
- D. Kim, K. Jeong, J. E. Kwon, H. Park, S. Lee, S. Kim and S. Y. Park, Nat. Commun., 2019, 10, 3089 CrossRef PubMed.
- Z. Xu, D. Gonzalez-Abradelo, J. Li, C. A. Strassert, B. J. Ravoo and D. S. Guo, Mater. Chem. Front., 2017, 1, 1847–1852 RSC.
- W. Zhou, J. Zhu, D. Fan, Y. Teng, X. Zhu and S. Dong, Adv. Funct. Mater., 2017, 27, 1704092 CrossRef.
- L. Hou, Y. Qin, J. Li, S. Qin, Y. Huang, T. Lin, L. Guo, F. Ye and S. Zhao, Biosens. Bioelectron., 2019, 143, 111605 CrossRef PubMed.
- S. A. Diaz, L. Giordano, T. M. Jovin and E. A. Jares-Erijman, Nano Lett., 2012, 12, 3537–3544 CrossRef CAS PubMed.
- S. A. Diaz, L. Giordano, J. C. Azcarate, T. M. Jovin and E. A. Jares-Erijman, J. Am. Chem. Soc., 2013, 135, 3208–3217 CrossRef CAS PubMed.
- M. Yu, P. Zhang, B. P. Krishnan, H. Wang, Y. Gao, S. Chen, R. Zeng, J. Cui and J. Chen, Adv. Funct. Mater., 2018, 28, 1804759 CrossRef.
- G. Naren, C. W. Hsu, S. M. Li, M. Morimoto, S. C. Tang, J. Hernando, G. Guirado, M. Irie, F. M. Raymo, H. Sundén and J. Andréasson, Nat. Commun., 2019, 10, 3996 CrossRef CAS PubMed.
- J. X. Bu, K. Watanabe, H. Hayasaka and K. Akagi, Nat. Commun., 2014, 5, 3799 CrossRef CAS.
- H. Wu, Y. Chen, X. Dai, P. Li, J. F. Stoddart and Y. Liu, J. Am. Chem. Soc., 2019, 141, 6583–6591 CrossRef CAS.
- C. W. Hsu, C. Sauvee, H. Sundén and J. Andréasson, Chem. Sci., 2018, 9, 8019–8023 RSC.
- M. Bälter, S. M. Li, M. Morimoto, S. C. Tang, J. Hernando, G. Guirado, M. Irie, F. M. Raymo and J. Andréasson, Chem. Sci., 2016, 7, 5867–5871 RSC.
- T. Wu, G. Zou, J. M. Hu and S. Y. Liu, Chem. Mater., 2009, 21, 3788–3798 CrossRef CAS.
- L. Y. Zhu, W. W. Wu, M. Q. Zhu, J. J. Han, J. K. Hurst and A. D. Q. Li, J. Am. Chem. Soc., 2007, 129, 3524–3526 CrossRef CAS PubMed.
- S. H. Lee, H. T. Bui, T. P. Vales, S. Cho and H. J. Kim, Dyes Pigm., 2017, 145, 216–221 CrossRef CAS.
- H. Wang, P. Zhang, B. P. Krishnan, M. Yu, J. Liu, M. Xue, S. Chen, R. Zeng, J. Cui and J. Chen, J. Mater. Chem. C, 2018, 6, 9897–9902 RSC.
- S. Wang, T. Li, X. Zhang, L. Ma, C. Li, X. Yao, D. Cao and X. Ma, ChemPhotoChem, 2019, 3, 568–574 CrossRef CAS.
- Z. Lin, H. Wang, M. Yu, X. Guo, C. Zhang, H. Deng, P. Zhang, S. Chen, R. Zeng, J. Cui and J. Chen, J. Mater. Chem. C, 2019, 7, 11515–11521 RSC.
- P. Deng, J. Sun, J. Chen, X. Zou and L. Liao, Carbohydr. Polym., 2019, 210, 379–388 CrossRef CAS.
- D. Kim, J. E. Kwon and S. Y. Park, Adv. Funct. Mater., 2018, 28, 1706213 CrossRef.
- S. A. Diaz, F. Gillanders, K. Susumu, E. Oh, I. L. Medintz and T. M. Jovin, Chem. – Eur. J., 2017, 23, 263–267 CrossRef CAS PubMed.
- S. Ishida, T. Fukaminato, D. Kitagawa, S. Kobatake, S. Kim, T. Ogata and S. Kurihara, Chem. Commun., 2017, 53, 8268–8271 RSC.
- M. Irie, T. Fukaminato, K. Matsuda and S. Kobatake, Chem. Rev., 2014, 114, 12174–12277 CrossRef CAS.
- K. Uno, H. Niikura, M. Morimoto, Y. Ishibashi, H. Miyasaka and M. Irie, J. Am. Chem. Soc., 2011, 133, 13558–13564 CrossRef CAS.
- M. Morimoto, Y. Takagi, K. Hioki, T. Nagasaka, H. Sotome, S. Ito, H. Miyasaka and M. Irie, Dyes Pigm., 2018, 153, 144–149 CrossRef CAS.
- R. Wang, S. Pu, G. Liu, S. Cui and W. Liu, J. Photochem. Photobiol., A, 2012, 243, 47–55 CrossRef CAS.
- K. Shibata, L. Kuroki, T. Fukaminato and M. Irie, Chem. Lett., 2008, 37, 832–833 CrossRef CAS.
- M. Irie, T. Lifka, K. Uchida, S. Kobatake and Y. Shindo, Chem. Commun., 1999, 747–750 RSC.
- C. C. Ko, W. H. Lam and V. Wing-Wah Yam, Chem. Commun., 2008, 5203–5205 RSC.
- R. Kanazawa, T. Nakashima and T. Kawai, J. Phys. Chem. A, 2017, 121, 1638–1646 CrossRef CAS.
- S. Huang, Z. Li, S. Li, J. Yin and S. Liu, Dyes Pigm., 2012, 92, 961–966 CrossRef CAS.
- M. T. W. Milder, J. L. Herek, J. Areephong, B. L. Feringa and W. R. Browne, J. Phys. Chem. A, 2009, 113, 7717–7724 CrossRef CAS PubMed.
- C. T. Poon, W. H. Lam, H. L. Wong and V. W. W. Yam, J. Am. Chem. Soc., 2010, 132, 13992–13993 CrossRef CAS PubMed.
- H. H. Liu and Y. Chen, J. Phys. Chem. A, 2009, 113, 5550–5553 CrossRef CAS PubMed.
- T. Fukaminato, S. Ishida and R. Metivier, NPG Asia Mater., 2018, 10, 859–881 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Synthesis, experimental section, and supporting figures. See DOI: 10.1039/c9cc10040g |
| ‡ There are examples of other DAE derivatives with similar or higher fluorescence quantum yields in the open isomeric form.32,38–44 In these compounds, however, the emission is originating from a distinct fluorophore associated with the photoswitch, and not integral part of the same. |
| This journal is © The Royal Society of Chemistry 2020 |