
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAn innovative approach for the synthesis of dual modality peptide imaging probes based on the native chemical ligation approach†
Ivan
Hawala
 a,
Lucia
De Rosa
b,
Silvio
Aime
a and
Luca Domenico
D’Andrea
*c
a,
Lucia
De Rosa
b,
Silvio
Aime
a and
Luca Domenico
D’Andrea
*c
aDipartimento di Biotecnologie Molecolari e Scienze per la Salute, Centro di Imaging Molecolare, Università degli Studi di Torino, Via Nizza 52, 10126, Torino (TO), Italy
bIstituto di Biostrutture e Bioimmagini, CNR, Via Mezzocannone 16, 80134 Napoli (NA), Italy
cIstituto di Biostrutture e Bioimmagini, CNR, Via Nizza 52, 10126 Torino (TO), Italy. E-mail: luca.dandrea@cnr.it
First published on 18th February 2020
Abstract
Peptide-targeting probes tagged with optical imaging and PET reporters may find applications in innovative diagnostic procedures and image-guided surgeries. The reported synthesis procedure is of general applicability to obtain dual imaging probes using fully unprotected moieties with a selective and rapid chemistry based on native chemical ligation.
Dual modality imaging probes are under intense scrutiny as they are useful tools for innovative diagnostic procedures and/or for monitoring therapeutic treatments.1 Dual-modality optical/PET probes allow for a better correlation between fluorescence and PET imaging experiments, especially when the structural difference between monomodality optical probes and the corresponding PET probes is associated with a not ignorable biological effect.2
Peptide-based probes3 are advantageous as they exhibit fast body clearance and an easy-to-perform quality control, when compared to the larger size proteins and antibodies. However, the preparation of a doubly labelled peptide probe usually requires a complex multi-step chemistry and the use of orthogonal protecting groups.4–6 Currently, the preparation of dual labelled peptides is often performed using solid phase techniques (Solid Phase Peptide Synthesis, SPPS) or by combining solid and solution phases. When the preparation is carried out in solution, one has to rely on the availability of properly designed chemoselective reactions. The solid phase approach requires the insertion of a molecule with at least two orthogonal protecting groups, compatible with the chemistry of the SPPS, and the use of a large excess of chelating and optical agents which should be stable to the SPPS and cleavage conditions. Alternatively, the peptide can be cleaved from the fully protected resin and, after purification, one or both agents can be coupled in solution upon selective deprotection, followed by the final peptide deprotection and purification. In some cases, it would be convenient to prepare a dual scaffold at first and then perform the bioconjugation in the solid or solution phase. In all cases, multiple synthesis and purification steps are required. To avoid laborious chemical procedures and the consumption of large excess of reagents, a limitation in the choice of imaging probes, chemoselective groups could be inserted for selective labelling in solution when working on fully deprotected peptides. Several chemoselective chemistries are available for specific bioconjugation steps such as those offered by the click chemistry and the chemical ligation approaches. For example, Sun and coworkers exploited a photo-click approach to efficiently generate dual PET/OI peptide imaging probes.2 However, despite the fact that dual-labelling probes can be obtained in only one bioconjugation step, this method has limitations due to the chemical nature and properties of the fluorophore moiety. Another interesting approach relies on the conjugation of peptides with strained internal alkynes for which the labelling with a base-catalyzed double addition of different imaging moieties modified with thiol functional groups was reported.7 In this case, the first thiol addition has to be carefully controlled to avoid undesired double labelling with the same moiety.
The use of the chemical ligation approach in the field of peptide chemistry has already found interesting applications, both in chemical syntheses and bioconjugation procedures.8–10 In particular, it has been stressed how chemical ligation approaches may provide advantageous strategies for protein and peptide site-specific modification.11–13
Native chemical ligation (NCL) was originally developed to generate long peptide chains (or proteins) while maintaining the native peptide backbone, which for many peptides/proteins is fundamental for preserving the bioactivity.14 This chemical approach allows for the coupling of two fully-deprotected peptide fragments in an aqueous solution to form an amide bond at the ligation site.15
In a typical NCL procedure, a C-terminal thioester peptide selectively reacts with an N-terminal cysteinyl peptide to afford a native polypeptide product (Fig. 1). No additional protecting groups are required for any of the amino acids found in the polypeptide, and, furthermore, the presence of internal cysteine residues does not hinder this reaction. For these reasons, thereby, NCL can be considered an invaluable regioselective reaction.16,17
 | ||
| Fig. 1 Mechanism of the native chemical ligation reaction. | ||
Herein, the utility of the NCL approach in the synthesis of dual optical/PET peptide probes is reported. This type of imaging probe may find application also in the field of image-guided surgery, where the PET tracer may act as a reporter of the targeting procedure in in vivo pre-surgery images and the fluorescent probe may guide the surgeon in the resection of the pathological lesion. Two well established tumour targeting peptides (AE10518 and cycloRGDfK19,20) were considered. First, they were derivatized with a cysteine residue to yield a and b compounds shown in Fig. 2. These molecules were then conjugated to the AAZTA chelator, preactivated as a MESNA thioester (c, in Fig. 2) via NCL. After the first NCL reaction was completed, the free thiol group of the Cys residue was exploited for pursuing the second site-specific labelling, via the thiol–maleimide chemistry21 to conjugate the maleimide fluorophore derivative (d, in Fig. 2).
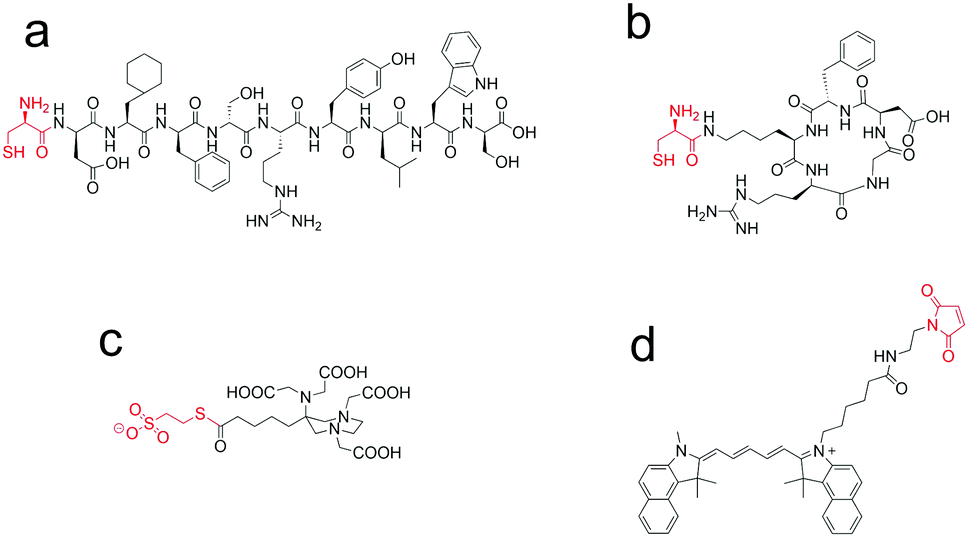 | ||
| Fig. 2 Chemical structures of the building blocks used in the procedure reported in Fig. 3: (a) Cys-AE105; (b) cycloRGDfK(C); (c) AAZTA-C4-CO-MES; (4) Cy 5.5 maleimide. | ||
The proposed approach can also be extended to protein-based probes, upon the insertion of a Cys residue at the protein N-terminal, thus paving a route to dual selectively labelled proteins.
Currently, the mainly used chelators such as 1,4,7,10-tetraazacyclododecane-1,4,7,10-tetraacetic acid (DOTA) have certain limitations,22 associated with the need for withstanding high temperatures for successful radiolabelling. In this work we used a polyaminopolycarboxylateheptadentate ligand based on a 1,4-diazepine scaffold (AAZTA) that has been thoroughly studied as a chelator for Gd3+ ions for MRI applications.23 This ligand has already been proven to form thermodynamically stable and kinetically inert gallium complexes24 and an AAZTA-RGD peptide conjugate was recently shown to complex 68Ga at room temperature in acetate buffer at pH 3.8.
As far as the fluorescent reporter is concerned, cy 5.5 was selected based on a large number of studies that have been carried out in the field of optical imaging using these molecules.25,26
The synthesis procedure is schematically depicted in Fig. 3. It starts from the thioester and 1,2 amino thiol components and consists of two reactions which, in principle, could be performed in sequence without the need for purification of the intermediate product. All reactions are performed in aqueous solution, using unprotected components.
 | ||
| Fig. 3 Synthesis scheme of the dual labelled peptide based probes (6 and 7, Fig. S1 and S2, ESI†): step (1) phosphate buffer pH 7.4, 5 M imidazole, 3 M guanidinium chloride, 50 mM TCEP, 1 h, RT; step (2) cy 5.5 maleimide, acetate pH 6.5, 3 h, RT. | ||
The peptides H-Cys(trt)-Asp(t-Bu)-Cha-Phe-D-Ser(t-Bu)-D-Arg(Pbf)-Tyr(t-Bu)-Leu-Trp(Boc)-Ser(t-Bu)-OH (1) (cysteinyl derivative of AE105 peptide, uPAR antagonist18) and cycloRGDfK(C) (2) (cysteinyl derivative of cycloRGDfK peptide, integrin antagonist27) were synthesized using the solid phase procedure. Then, they were fully deprotected and characterized (Fig. S3, ESI†). (tBu)4-AAZTA-C4-COOH was preactivated as a thioester with MESNA and then totally deprotected with a mixture of TFA/TIS/H2O (compound 3, Fig. S4 and S5, ESI†), to allow the subsequent condensation with 1 and 2, using the NCL approach (Fig. 3, step 1). The reaction between 1 or 2 and 3 was performed in 0.2 M phosphate buffer medium (pH 7.4) supplemented with 5 M imidazole, 3 M guanidinium chloride and 50 mM TCEP. Guanidinium chloride improves the solubility of the starting materials; the increased concentration of the reagents allows the decrease of either the reaction time or the reaction volume. Imidazole was used instead of a thiol (e.g. 4-mercaptophenylacetic acid) as a NCL reaction catalyst,28 and TCEP was added to the mixture as a reducing agent with the purpose of getting the full availability of the thiol group of the cysteine residue, avoiding the formation of disulfide bonds that would hinder the NCL reaction. After the transthioesterification step, an intramolecular rearrangement gives a stable amide bond between the N-terminal amino group of the cysteine and the carboxylic group of the externally protruded arm of AAZTA-C4-COOH. Overall the NCL procedure was completed in about 30–60 min yielding, after RP-HPLC purification, a highly pure product (Fig. S6, ESI†).
The transposition of the chemical bond from the side chain thiol group to the α-amine group made the thiol group of the cysteine available for the subsequent chemoselective reaction with the maleimide pre-activated fluorophore (i.e. cy 5.5) (Fig. 3, step 2). For this step, maleimide–thiol chemistry was used, as maleimide-derivatized fluorophores are commercially available. Of course, other thiol selective chemistry can also be exploited. The final reaction between the free cysteine thiol and the maleimide moiety of the pre-activated fluorophore, carried out in aqueous solution at almost neutral pH, was completed within a few hours at room temperature (Fig. S7, ESI†).
AAZTA-C4-CO-Cys(Cy5.5)-AE105 (6) was synthesized with an overall yield of 24% and a chemical purity of 95%. AAZTA-C4-CO-(Cy 5.5)-cycloRGDfK(C) (7) was synthesized with an overall yield of 6.4% and a chemical purity of 95%. The low values of the overall yields are mainly related to a low recovery after the purification steps, as bioconjugation reactions proceeded almost to completion as assessed by RP-HPLC analyses.
We also performed the synthesis of 6 and 7 in two consecutive steps by avoiding the purification of the species 4 and 5. It was carried out by adding cy 5.5 maleimide to the reaction mixture of the chemical ligation step after having checked the full conversion of the reagent by RP-UPLC. However, the free thiol-containing anion, released during the NCL reaction between AAZTA thioester and the peptides, can react with the fluorophore–maleimide thus reducing the part of the reagent needed for the second labelling reaction. Unfortunately, the side product formed from MES/cy 5.5 maleimide had a similar HPLC retention time to the desired product 6. This led to obvious purification inconveniences. In summary, the latter synthesis approach, in the case of the synthesis of 6, gave a similar overall yield of 22% (it was 24% for the former approach). Conversely, the overall yield increased in the case of the synthesis of 7 passing from 6.4% to 23% highlighting the influence of the purification step on the overall yield. In both cases, the final product was obtained with a chemical purity of 95%.
The AAZTA ligand on 6 and 7 yielded the respective Ga(III) complexes by mixing equimolar amounts of the peptide-containing probe and GaCl3 in a 50![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :50 solution of 0.1 M acetate buffer pH (3.8)/acetonitrile medium, at room temperature for 10 minutes under magnetic stirring. The reactions were monitored by UPLC-MS (8 and 9, Fig. S8, ESI†). The final products were characterized by UV-Vis spectroscopy. UV-Vis spectra of 8 and 9 showed, according to the parent cy 5.5 maleimide spectrum, λmax at 692 nm, suggesting the preservation of the intact chromophore moiety in the final molecule (Fig. 4). The concentrations of 8 and 9 complexes were quantified by UV-Vis spectroscopy and used as such for the in vitro targeting experiments, on the assumption that tiny amounts of acetonitrile (<0.1% v/v) do not compromise the viability of the cells during the experiment time.
:50 solution of 0.1 M acetate buffer pH (3.8)/acetonitrile medium, at room temperature for 10 minutes under magnetic stirring. The reactions were monitored by UPLC-MS (8 and 9, Fig. S8, ESI†). The final products were characterized by UV-Vis spectroscopy. UV-Vis spectra of 8 and 9 showed, according to the parent cy 5.5 maleimide spectrum, λmax at 692 nm, suggesting the preservation of the intact chromophore moiety in the final molecule (Fig. 4). The concentrations of 8 and 9 complexes were quantified by UV-Vis spectroscopy and used as such for the in vitro targeting experiments, on the assumption that tiny amounts of acetonitrile (<0.1% v/v) do not compromise the viability of the cells during the experiment time.
 | ||
| Fig. 4 UV-Vis spectra of cy 5.5 maleimide, compound 8 and compound 9. | ||
Human glioblastoma U-87 MG cells overexpressing either uPAR or αvβ3 integrin receptors were used to assess the binding properties of the synthesized dual probes (compound 8 and 9) by flow cytometry. Tumour cells (105 per well) were incubated in a medium containing different concentrations of 8 or 9 (from 0 to 0.4 μg) for 30 minutes at 273 K. The representative results from flow cytometry experiments with Ga-AAZTA-C4-CO-Cys(Cy5.5)-AE105 (8) and Ga-AAZTA-C4-CO-(Cy5.5)-cycloRGDfK(C) (9) are shown in Fig. S9 and S10 (ESI†). Both dual probes were able to bind to U-87 MG cells. Furthermore, both probes showed a dose-dependent behaviour (Fig. 5a and b). Overall, these in vitro experiments demonstrate that the dual-labelled peptides are able to interact with uPAR or integrin receptors.29
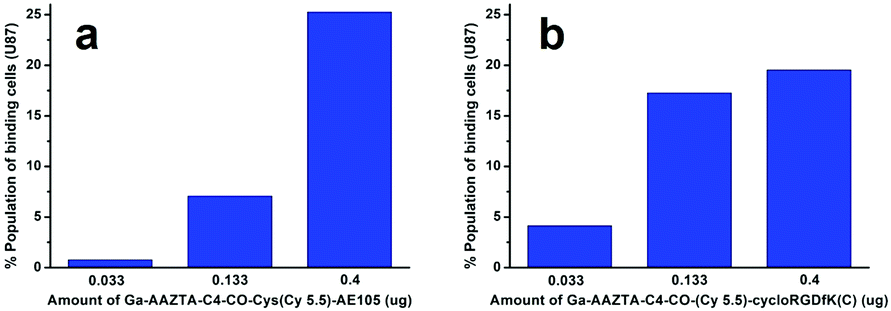 | ||
| Fig. 5 (a) Histogram plot of U-87 MG cells (105) analysed at FACS after incubation for 1 h at 273 K with 0.033, 0.133 and 0.400 μg of Ga-AAZTA-C4-CO-Cys(Cy5.5)-AE105 (8); (b) histogram plot of U-87 MG cells (105) analysed after incubation for 1 h at 273 K with 0.033, 0.133 and 0.400 μg of Ga-AAZTA-C4-CO-(Cy5.5)-cycloRGDfK(C) (9). | ||
The reported synthesis procedure allows for the preparation of a dual labelled peptide based probe. Using two chemoselective reactions, the fluorescent and the chelating moieties are inserted at site-specific positions without the use of protecting groups. The targeting peptide is modified with the introduction of a cysteine residue, which can be easily inserted during peptide synthesis, minimizing the impact on the overall molecular structure even in the case of short peptide sequences. All the bioconjugation reactions are performed in aqueous solution, under mild conditions and using unprotected reagents, suggesting that an analogous procedure could be easily translated to the preparation of protein-based probes, not presenting exposed Cys residue, through the introduction of an N-terminal cysteine.
In conclusion, the herein reported results show that a synthesis strategy can be set-up to obtain peptide-containing molecules (e.g. AE105 or cycloRGDfK) functionalized with a fluorophore (e.g. cy 5.5) and a chelator (e.g. AAZTA). The reported synthesis procedure can be generally applied to obtain dual imaging probes. Compounds 6 and 7 if labelled with Ga-68 could be considered as candidates for dual detention with PET and optical imaging. Such probes might be particularly useful in image-guided surgery via the detention of the fluorescence reporters once the occurrence of successful targeting has been verified pre-surgery by the acquisition of PET images.
I. H. is grateful to Bracco Imaging Spa for financial support of the PhD program. The authors are very grateful to Marta Tapparo (University of Turin, Italy) for her precious support during the in vitro experiment evaluations. L. D. R. was supported by “Fondazione Umberto Veronesi”.
Conflicts of interest
There are no conflicts to declare.References
- A. Louie, Chem. Rev., 2010, 110, 3146–3195 CrossRef CAS
.
- L. Sun, J. Ding, W. Xing, Y. Gai, J. Sheng and D. Zeng, Bioconjugate Chem., 2016, 27, 1200–1204 CrossRef CAS PubMed
- S. Mizukami, R. Takikawa, F. Sugihara, M. Shirakawa and K. Kikuchi, Angew. Chem., Int. Ed., 2009, 48, 3641–3643 CrossRef CAS PubMed
- C. Li, W. Wang, Q. Wu, S. Ke, J. Houston, E. Sevick-Muraca, L. Dong, D. Chow, C. Charnsangavej and J. G. Gelovani, Nucl. Med. Biol., 2006, 33, 349–358 CrossRef CAS PubMed
- H. Xu, K. Baidoo, A. J. Gunn, C. A. Boswell, D. E. Milenic, P. L. Choyke and M. W. Brechbiel, J. Med. Chem., 2007, 50, 4759–4765 CrossRef CAS PubMed
- K. R. Bhushan, P. Misra, F. Liu, S. Mathur, R. E. Lenkinski and J. V. Frangioni, J. Am. Chem. Soc., 2008, 130, 17648–17649 CrossRef CAS PubMed
- Y. Sun, X. Ma, K. Cheng, B. Wu, J. Duan, H. Chen, L. Bu, R. Zhang, X. Hu, Z. Deng, L. Xing, X. Hong and Z. Cheng, Angew. Chem., Int. Ed., 2015, 54, 5981–5984 CrossRef CAS
-
L. D. D'Andrea and A. Romanelli, Chemical Ligation: Tools for Biomolecule Synthesis and Modification, Wiley, 2017 Search PubMed
-
W. R. Algar, P. Dawson and I. L. Medintz, Chemoselective and Bioorthogonal Ligation Reactions. Concepts and Applications, Wiley, 2017 Search PubMed
- L. De Rosa, A. Russomanno, A. Romanelli and L. D. D'Andrea, Molecules, 2013, 18, 440–465 CrossRef CAS PubMed
- L. De Rosa, A. L. Cortajarena, A. Romanelli, L. Regan and L. D. D'Andrea, Org. Biomol. Chem., 2012, 10, 273–280 RSC
- C. P. Hackenberger and D. Schwarzer, Angew. Chem., Int. Ed., 2008, 47, 10030–10074 CrossRef CAS PubMed
- L. De Rosa, R. Di Stasi, L. Longhitano and L. D. D'Andrea, Bioorg. Chem., 2019, 91, 103160 CrossRef CAS PubMed
- P. E. Dawson, T. W. Muir, I. Clark-Lewis and S. B. Kent, Science, 1994, 266, 776–779 CrossRef CAS PubMed
- S. Chandrudu, P. Simerska and I. Toth, Molecules, 2013, 18, 4373–4388 CrossRef CAS PubMed
- S. Kent, Y. Sohma, S. Liu, D. Bang, B. Pentelute and K. Mandal, J. Pept. Sci., 2012, 18, 428–436 CrossRef CAS PubMed
- T. M. Hackeng, C. M. Mounier, C. Bon, P. E. Dawson, J. H. Griffin and S. B. Kent, Proc. Natl. Acad. Sci. U. S. A., 1997, 94, 7845–7850 CrossRef CAS PubMed
- Z. B. Li, G. Niu, H. Wang, L. He, L. Yang, M. Ploug and X. Chen, Clin. Cancer Res., 2008, 14, 4758–4766 CrossRef CAS PubMed
- R. Haubner, R. Gratias, B. Diefenbach, S. L. Goodman, A. Jonczyk and H. Kessler, J. Am. Chem. Soc., 1996, 118, 7461–7472 CrossRef CAS
- H. P. Hammes, M. Brownlee, A. Jonczyk, A. Sutter and K. T. Preissner, Nat. Med., 1996, 2, 529–533 CrossRef CAS PubMed
- B. H. Northrop, S. H. Frayne and U. Choudhary, Polym. Chem., 2015, 6, 3415–3430 RSC
- E. W. Price and C. Orvig, Chem. Soc. Rev., 2014, 43, 260–290 RSC
- L. Manzoni, L. Belvisi, D. Arosio, M. P. Bartolomeo, A. Bianchi, C. Brioschi, F. Buonsanti, C. Cabella, C. Casagrande, M. Civera, M. De Matteo, L. Fugazza, L. Lattuada, F. Maisano, L. Miragoli, C. Neira, M. Pilkington-Miksa and C. Scolastico, ChemMedChem, 2012, 7, 1084–1093 CrossRef CAS PubMed
- Z. Baranyai, F. Uggeri, A. Maiocchi, G. B. Giovenzana, C. Cavallotti, A. Takàcs, I. Toth, I. Benyei, E. Brucher and S. Aime, Eur. J. Inorg. Chem., 2013, 147–162 CrossRef CAS
- Y. Shimizu, T. Temma, I. Hara, R. Yamahara, E. Ozeki, M. Ono and H. Saji, J. Fluoresc., 2012, 22, 719–727 CrossRef CAS PubMed
- V. Ntziachristos, J. Ripoll, L. V. Wang and R. Weissleder, Nat. Biotechnol., 2005, 23, 313–320 CrossRef CAS PubMed
- M. A. Russo, M. Paolillo, Y. Sanchez-Hernandez, D. Curti, E. Ciusani, M. Serra, L. Colombo and S. Schinelli, Int. J. Oncol., 2013, 42, 83–92 CrossRef CAS PubMed
- K. Sakamoto, S. Tsuda, M. Mochizuki, Y. Nohara, H. Nishio and T. Yoshiya, Chem. – Eur. J., 2016, 22, 17940–17944 CrossRef CAS PubMed
- K. Chen, L. P. Yap, R. Park, X. Hui, K. Wu, D. Fan, X. Chen and P. S. Conti, Amino Acids, 2012, 42, 1329–1337 CrossRef CAS PubMed
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9cc09980h |
| This journal is © The Royal Society of Chemistry 2020 |