
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceKeto–enol tautomerization drives the self-assembly of leucoquinizarin on Au(111)†
Roberto
Costantini
 ab,
Luciano
Colazzo
cd,
Laura
Batini
e,
Matus
Stredansky
ab,
Mohammed S. G.
Mohammed
cd,
Simona
Achilli
e,
Luca
Floreano
b,
Guido
Fratesi
e,
Dimas G.
de Oteyza
cdf and
Albano
Cossaro
*b
ab,
Luciano
Colazzo
cd,
Laura
Batini
e,
Matus
Stredansky
ab,
Mohammed S. G.
Mohammed
cd,
Simona
Achilli
e,
Luca
Floreano
b,
Guido
Fratesi
e,
Dimas G.
de Oteyza
cdf and
Albano
Cossaro
*b
aPhysics Department of University of Trietse, via A. Valerio 2, 34127 Trieste, Italy
bCNR-IOM, Area Science Park, Strada Statale 14, km 163,5, 34149 Trieste, Italy. E-mail: cossaro@iom.cnr.it
cDonostia International Physics Center, Paseo Manuel Lardizabal 4, E-20018 Donostia-San Sebastián, Spain
dCentro de Física de Materiales (CSIC-UPV/EHU) – MPC, Paseo Manuel de Lardizabal, 5 – E-20018 Donostia-San Sebastián, Spain
eDipartimento di Fisica “Aldo Pontremoli”, Università degli Studi di Milano, Milano, Italy
fIkerbasque, Basque Foundation for Science, E-48011 Bilbao, Spain
First published on 6th February 2020
Abstract
The self-assembly of leucoquinizarin molecules on Au(111) surfaces is shown to be characterized by the molecules mostly being in their keto–enolic tautomeric form, with evidence of their temporary switching to other tautomeric forms. This reveals a metastable chemistry of the assembled molecules, to be considered for their possible employment in the formation of more complex hetero-organic interfaces.
Hydroxyl or oxygen containing derivatives of anthracene are an important class of molecules, widely employed in industrial processes such as dye production,1,2 drugs synthesis,3 and others.4 They are characterized by a fast redox response, which makes them convenient building blocks for the design of cathodes of a new generation of batteries5–7 and for the synthesis of complex hybrid interfaces for photovoltaic devices.8 Their color and in general their electronic properties can be finely tailored by varying the functionalization of the aromatic body.9 The presence of one or more oxygen terminations promotes the occurrence of keto–enol tautomerism in these compounds, which can be of relevant importance for their functionality. For instance, in complexes formed by crown ethers and anthracene derivatives, the ion complexation of the former acts as a switch of the tautomerism of the anthracene group and promotes its luminescence, making the complexes suitable for the design of ion sensors.10–13 As a matter of fact, most of the employed anthracene derivatives are based on 9,10-anthraquinone, an anthracene with double bonded oxygen atoms substituting the 9,10 positions. We report here a combined imaging, spectroscopic and theoretical study of the assembly on Au(111) surfaces of leucoquinizarin (LQZ, C14H10O4), an anthracene with four hydroxyl groups in positions 1, 9, 4, and 10 (Fig. 1), adopted in the chemical industry for the synthesis of dyes. The molecule goes through keto–enol tautomerism and some of the tautomers are represented in Fig. 1. The presence of pairs of hydroxyl groups positioned in a double diol configuration makes LQZ a potential building block for extended covalent networks. In particular, one could exploit its tendency toward aldol condensation with hydroxyl molecules,14 as well as its covalent coupling with boronic molecules upon a diol-boronic condensation reaction.15 The latter perspective would be of interest due to the increasing importance that boronic chemistry has gained in recent years as a tool for the synthesis of complex 2D interfaces.16–18 In the view of exploring these synthesis routes to possibly exploit the electronic and optical properties of anthraquinone derivatives in more extended architectures, the chemistry of LQZ on surfaces needs to be properly investigated first. Both experimental19 and theoretical20 findings indicate the T1 configuration as the most stable among the possible tautomers depicted in Fig. 1, both in the gas phase and in solution. Our findings support T1 to be the LQZ form also on the Au(111) surface. The room temperature growth of the LQZ sub-monolayer is characterized by differently ordered assemblies. The imaging of the most ordered phase is reported in Fig. 2 and exhibits a molecular assembly characterized by the presence of chiral pores, spaced by 2.5 nm. The morphology resembles the giant honeycomb architecture found for the anthraquinone molecule on Cu(111).21 Here however, the presence of additional oxygen terminations with respect to anthraquinone likely promotes a more compact assembly. Domains with both chiralities are observed on the same surface. The supramolecular structure corresponds to a hierarchical assembly with molecular trimers as building units, which remain together upon controlled molecular manipulation with the scanning probe (Fig. 2b and c). As already mentioned, the ordered assembly of Fig. 2, observed for low coverage (<0.5 ML), is not the only morphology obtained in the preparations. At higher coverage, or with larger deposition rate, the nanopore domains become less regular due to an increase of the molecular density, finally yielding a disordered phase at completion of the first layer (ESI,† Fig. S1). Interestingly, molecular trimers, even though characterized by a less regular geometry than those reported in Fig. 2, can still be identified as the recurring motif of the architectures. High resolution imaging with a CO functionalized tip is shown in Fig. 2d and allows observation of more details of the LQZ assembly and of the intermolecular interactions. All the molecules display the same aspect: one bright hexagonal carbon ring at one end, a darker and more irregular hexagonal ring in the center, and an even darker and strongly deformed structure at the opposite end. We performed DFT calculations in order to have better insight into both the assembly and the electronic properties of the LQZ molecule. The details of the calculations and of the resulting structures are reported in the ESI.† Our findings confirm the conformer T1 as the most stable form, with the purely hydroxylic T0 having 0.9 eV higher total energy in the gas phase. Moreover, the diketo form of the terminal quinol of T1 introduces a distortion of the anthracene backbone. This is likely the cause of the blurred aspect of one of the molecule terminations as imaged in Fig. 2d and suggests an assembly scheme with the aromatic ring (opposite to the quinol ring) pointing towards the pores. The superimposed molecular model in Fig. 2d is the result of DFT relaxation of a free-standing LQZ layer, as obtained starting from the structure suggested by STM. The analysis of the intermolecular bonding confirms that the oxygen atoms have a determinant role in the assembly. In particular, the interaction between the keto–enolic terminations of molecules belonging to adjacent trimers drives the assembly of the porous architecture (see the ESI† for details).
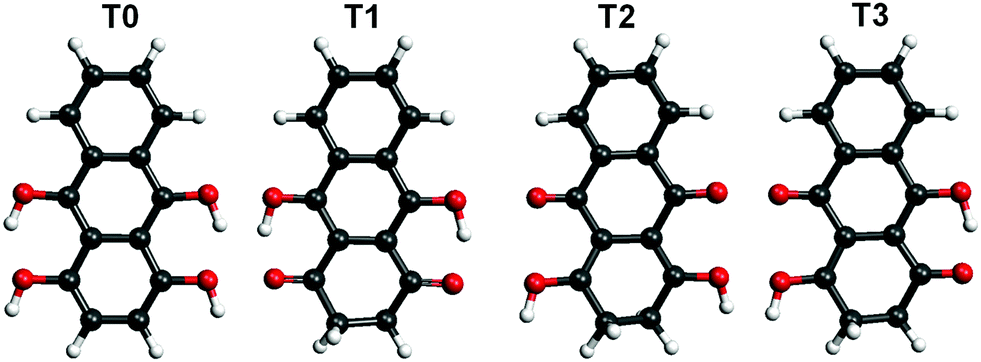 | ||
| Fig. 1 Molecular structure of four tautomers of the LQZ molecule. | ||
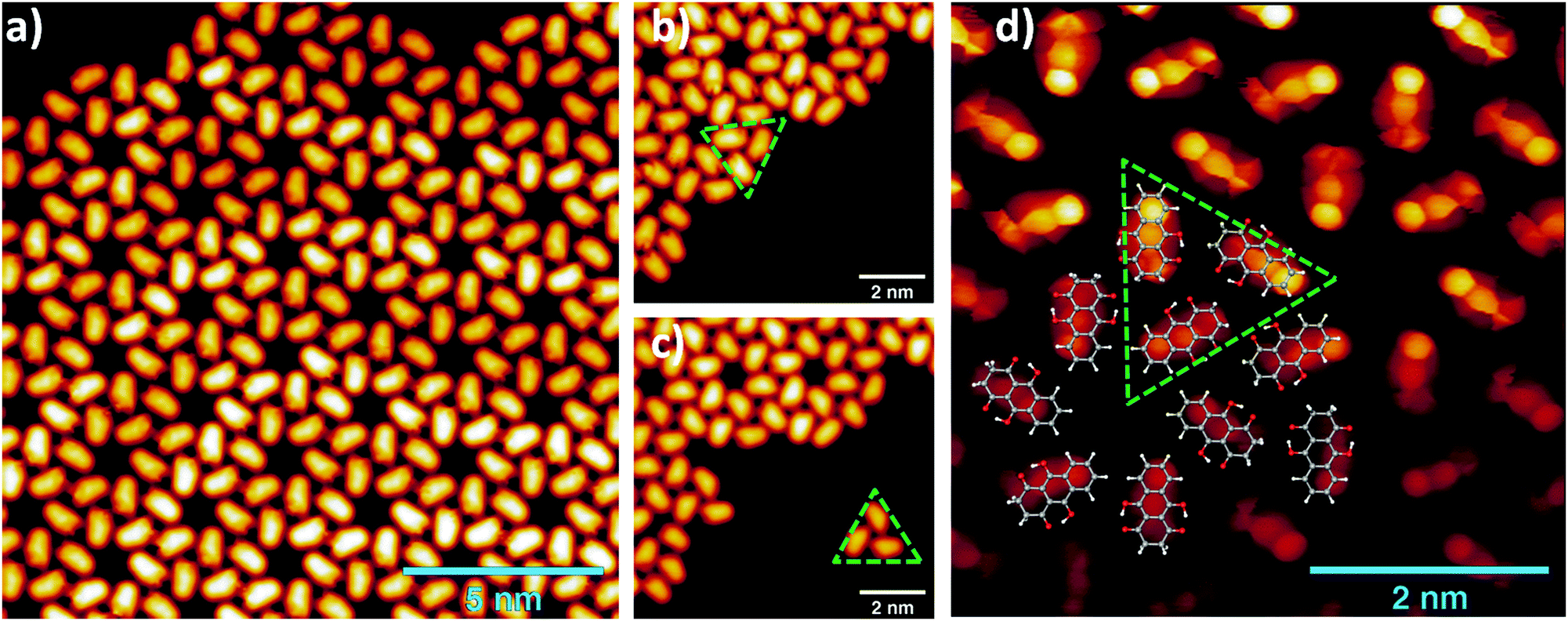 | ||
| Fig. 2 (a) STM images of a LQZ submonolayer showing the presence of nanometric pores (0.05 V, 100 pA). (b and c) Tip manipulation allows the separation of a trimer from the assembly, which is identified as the building block of the architecture (0.1 V, 50 pA). (d) Bond resolution image reveals a distorted termination of the LQZ molecules, which can be related to the diketo phenol of the T1 tautomer. The superimposed overlayer model results from the DFT calculations of the assembly, obtained as detailed in the ESI.† | ||
A further important aspect of LQZ assemblies, observed by STM measurements, is their unstable imaging. This is shown by way of example in Fig. 3. Panel (b) displays the image of a disordered chain of molecules, measured on a low coverage LQZ layer deposited on Au(111) held at 120 K. Maintaining the probe at the point marked by the blue cross and monitoring the current under open feedback conditions provides a trace as shown in panel (a), characterized by the presence of telegraph noise that is absent on the bare surface. By comparison with similar observations in previous studies, this effect can be related to the switching of the molecule to different tautomeric forms,22–24 although switching to different adsorption configurations cannot be excluded either. The curve exhibits two switching levels beside the most stable one, as evidenced by the blue dotted lines in the graph.
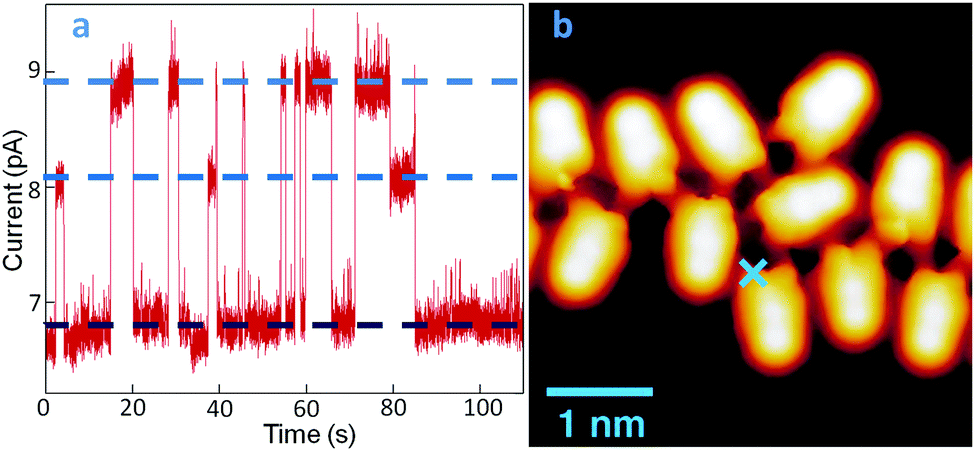 | ||
| Fig. 3 (a) Tunneling current measured in correspondence of the cross indicated in the image (panel b, −0.15 V, 1 nA) of a very low coverage LQZ layer, as a function of the measuring time. Three different current levels are measured, suggesting a dynamics of the LQZ chemistry in the assembly, possibly due to tautomeric switching. | ||
However, the switching rate and the different level's relative dominance greatly vary as the tip is positioned differently (Fig. S4, ESI†). While this proves the molecule-related origin of the switching, it also reveals the influence of the location-dependent tip-molecule interactions, in line with other well-known tautomerization switches revealed in porphycenes.24 Another similarity between the two systems is the promotion of the switching events by vibrational excitations of the molecule, which becomes obvious from the notably increased instabilities at bias values above ±50 meV, and a further increase at around ±350 meV. The symmetric thresholds for both bias polarities are a fingerprint of inelastic tunneling processes that we associate, in the absence of electronic orbitals and transitions at these low energies, to molecular vibrational modes. It should be noted, however, that instabilities in the tunneling gap are observed even at 1 meV (Fig. 3a), whereby tip-induced effects driven by inelastic tunneling excitations are minimized. The DFT energy difference between T1 and the other tautomers, as calculated for a free molecule, is too large for a tautomeric switch to be at the origin of the tunneling current behavior presented in Fig. 3, which has been measured at 4.3 K. However, it has to be considered that experimentally, both the intermolecular hydrogen bonding and the interaction with the substrate can considerably lower this energy barrier. Moreover, transitions to intermediate tautomers, where only one of the enolic terminations of T1 has switched, can be considered at the origin of the telegraph noise, with consequent reduced effort in terms of energy barrier.
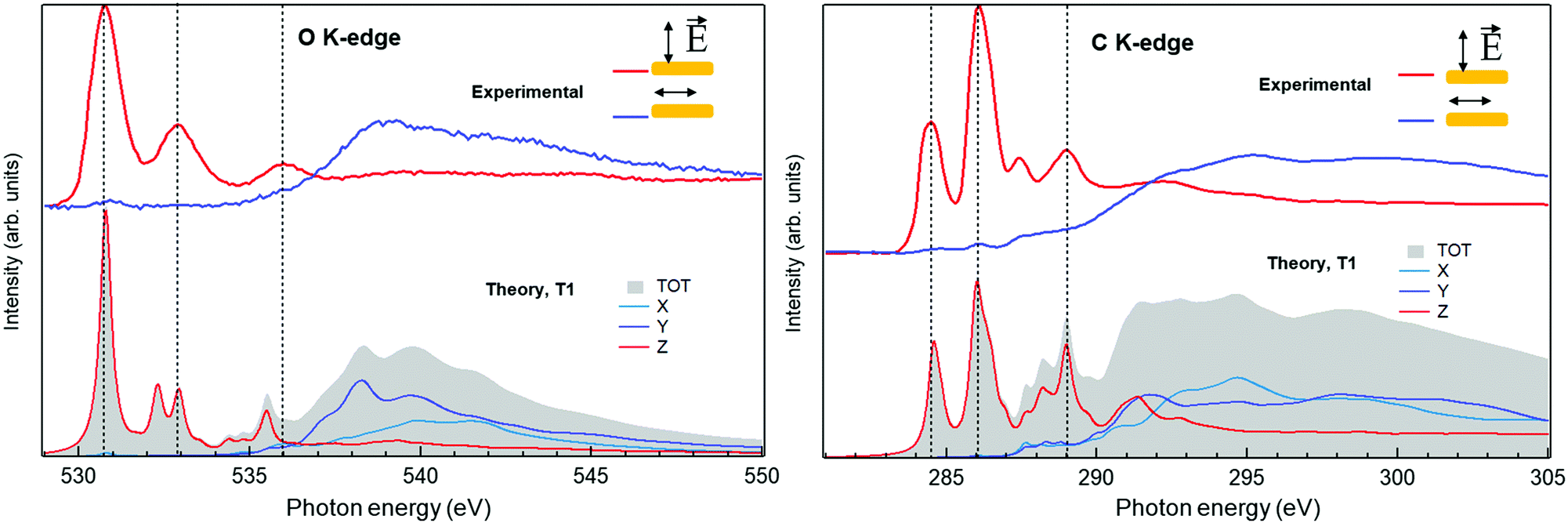
To further investigate the chemistry and the morphology of the system we performed X-ray spectroscopy measurements at the ALOISA beamline25 and at its ANCHOR-SUNDYN endstation26 at the Elettra Synchrotron. Fig. 4 reports the O and C K-edge NEXAFS spectra taken on an LQZ monolayer. The carbon spectra resemble the anthracene ones,27 apart from the intensity ratio between the first two resonances, at around 285 eV. Both edges present a strong dichroism, with the lower energy transitions having maximum intensity when the polarization of the electric field is perpendicular to the surface. We calculated the eigenfunctions for the ground state of the four tautomers, as well as the density of states projected onto states of π and σ symmetry. The four conformers exhibit the same symmetry of electronic states around the HOMO. In particular, the HOMO, LUMO and LUMO+1 are π states, whereas σ-symmetry can be found for the HOMO−1 (see the ESI†). The almost perfect dichroism indicates therefore that molecules are adsorbed flat on the surface and that neither the interaction with the substrate, nor the hydrogen bonding scheme the molecules are involved in, introduce relevant distortion of the electronic structure. The lower panels of Fig. 4 report the calculated spectra for the T1 tautomer, which provides a better match to the data than other configurations. Photoemission spectroscopy (XPS) also corroborates the presence of T1 species on the surface. The O1s and C1s peaks are shown in Fig. 5, as measured on an LQZ monolayer, and as calculated for the T1 molecule. No significant differences have been found in the XPS profiles taken on films with lower coverage. The O1s peak presents two broad components, of the same intensity, found at 531.3 eV and 532.7 eV, which can be assigned to carbonyl (![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O) and to hydroxyl (–OH) oxygen species respectively.28 The profile of C1s is more complex and can be fitted by five components. The calculated spectra for T1 nicely reproduce the experimental profiles and confirm the O1s assignment. Regarding the C1s peak, the highest energy component of C1s at ∼289 eV is not reproduced by calculations and can be assigned to energy loss due to inner excitations of the molecule. The C1s components 3 and 4 are due to the carbon atoms linked to the hydroxyl and carbonyl oxygen atoms respectively. We ascribe both the broadness of the O1s components and the minor discrepancies between experimental and calculated C1s profiles to the intermolecular bonds that are not considered by the calculation, as well as to the morphologic disorder of the assembly and to the possible presence of different tautomers on the surface. In this regard, we remark that, the X-ray spectroscopy measurements have been taken at room temperature, whereas STM has been performed at 4 K. We can speculate that these conditions promote a relevant portion of LQZ molecules in a tautomeric form different from T1.
O) and to hydroxyl (–OH) oxygen species respectively.28 The profile of C1s is more complex and can be fitted by five components. The calculated spectra for T1 nicely reproduce the experimental profiles and confirm the O1s assignment. Regarding the C1s peak, the highest energy component of C1s at ∼289 eV is not reproduced by calculations and can be assigned to energy loss due to inner excitations of the molecule. The C1s components 3 and 4 are due to the carbon atoms linked to the hydroxyl and carbonyl oxygen atoms respectively. We ascribe both the broadness of the O1s components and the minor discrepancies between experimental and calculated C1s profiles to the intermolecular bonds that are not considered by the calculation, as well as to the morphologic disorder of the assembly and to the possible presence of different tautomers on the surface. In this regard, we remark that, the X-ray spectroscopy measurements have been taken at room temperature, whereas STM has been performed at 4 K. We can speculate that these conditions promote a relevant portion of LQZ molecules in a tautomeric form different from T1.
 | ||
| Fig. 4 O1s and C1s XPS spectra measured on the LQZ monolayer (top panels) and simulated for the T1 tautomer (bottom). | ||
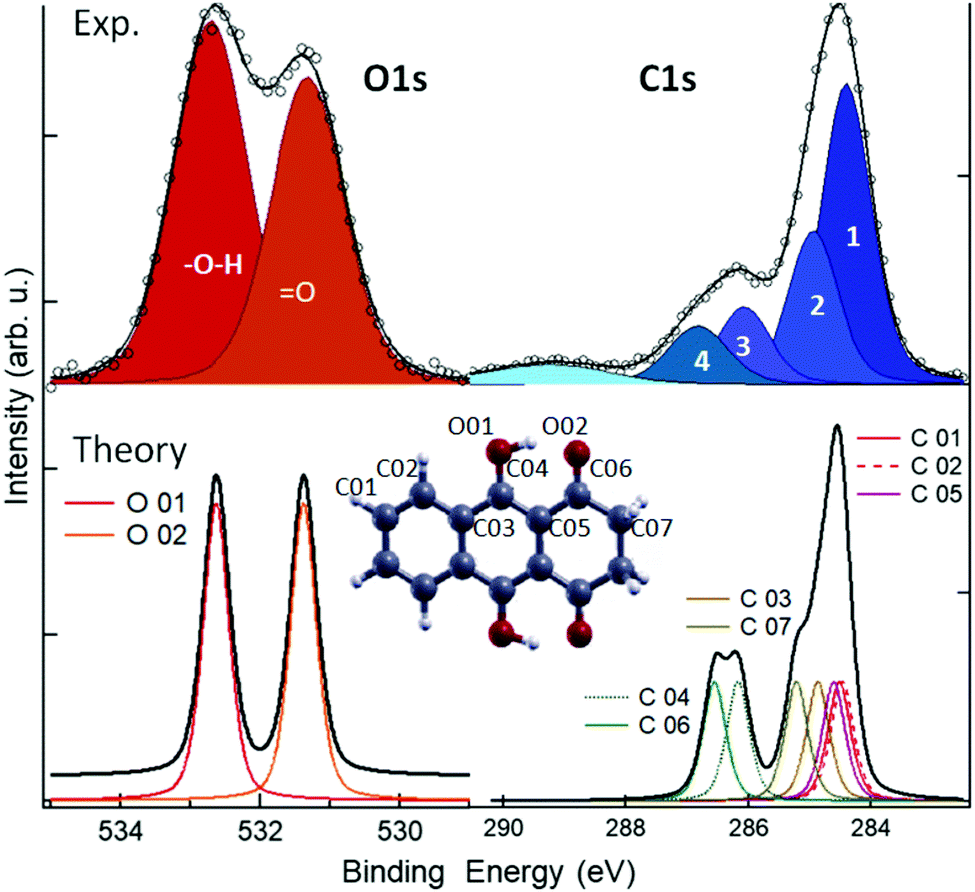 | ||
| Fig. 5 XPS spectra measured (top) and calculated for the T1 molecule (bottom). See the ESI,† for the comparison with the calculations of the other tautomers. | ||
In conclusion, the assembly of LQZ molecules on Au(111) has been characterized in terms of the morphology and of the electronic properties of the supramolecular architecture. A trimer of hydrogen bonded molecules has been identified as a recurring motif of the assembly and as a building block of differently ordered superstructures. The assembly scheme that can be inferred from the STM images on the highly ordered structures is consistent with the LQZ molecules predominantly in their keto–enolic tautomeric form. The same configuration is compatible with the X-ray spectroscopy results, as simulated by DFT calculations. We ascribe a certain degree of unpredictability of the morphology, as the molecular density increases, to the tautomeric switch the molecules can experience on the surface. This is corroborated by the observation of telegraph noise in the STS measurements. As different tautomers present different chemical properties of their oxygen terminations, our findings have to be taken into account in the perspective of employing LQZ as a building block of more complex interfaces and of the possible on-surface reactions it can be involved in.
We acknowledge: the CINECA award under the ISCRA initiative (Grant No. HP10CB0ZW2), the MIUR SIR grant SUNDYN (RBSI14G7TL, CUP B82I15000910001), and funding from the European Union's Horizon 2020 programme (Grant Agreement No. 635919) and from the Spanish MINECO (Grant No. MAT2016-78293-C6).
Conflicts of interest
There are no conflicts to declare.Notes and references
- D. R. Waring and G. Hallas, The Chemistry and application of dyes, Plenum Press, 1990 Search PubMed.
- K. Hunger, Industrial dyes: chemistry, properties, applications, Wiley-VCH, 2003 Search PubMed.
- J. William Lown, Pharmacol. Ther., 1993, 60, 185–214 CrossRef.
- G. Diaz-Muñoz, I. L. Miranda, S. K. Sartori, D. C. de Rezende and M. A. N. Diaz, Studies in Natural Products Chemistry, 2018 Search PubMed.
- Z. Song, H. Zhan and Y. Zhou, Chem. Commun., 2009, 448–450 RSC.
- W. Wang, W. Xu, L. Cosimbescu, D. Choi, L. Li and Z. Yang, Chem. Commun., 2012, 48, 6669–6671 RSC.
- B. Huskinson, M. P. Marshak, C. Suh, S. Er, M. R. Gerhardt, C. J. Galvin, X. Chen, A. Aspuru-Guzik, R. G. Gordon and M. J. Aziz, Nature, 2014, 505, 195–198 CrossRef CAS PubMed.
- M. Castelaın, H. J. Salavagione, R. Gomez and J. Luis Segura, Chem. Commun., 2011, 47, 7677–7679 RSC.
- D. Jacquemin, J. Preat, M. Charlot, V. Wathelet, J. M. André and E. A. Perpète, J. Chem. Phys., 2004, 121, 1736–1743 CrossRef CAS PubMed.
- M. Alaparthi, K. Mariappan, E. Dufek, M. Hoffman and A. G. Sykes, RSC Adv., 2016, 6, 11295–11302 RSC.
- P. N. Basa, A. Bhowmick, L. M. Horn and A. G. Sykes, Org. Lett., 2012, 14, 2698–2701 CrossRef CAS PubMed.
- M. Kadarkaraisamy and A. G. Sykes, Inorg. Chem., 2006, 45, 779–786 CrossRef CAS PubMed.
- B. Kampmann, Y. Lian, K. L. Klinkel, P. A. Vecchi, H. L. Quiring, C. C. Soh and A. G. Sykes, J. Org. Chem., 2002, 67, 3878–3883 CrossRef CAS.
- C. E. Lewis, J. Org. Chem., 1970, 35, 2938–2942 CrossRef CAS.
- Y. Furikado, T. Nagahata, T. Okamoto, T. Sugaya, S. Iwatsuki, M. Inamo, H. D. Takagi, A. Odani and K. Ishihara, Chem. – Eur. J., 2014, 20, 13194–13202 CrossRef CAS PubMed.
- S. Clair, M. Abel and L. Porte, Chem. Commun., 2014, 50, 9627–9635 RSC.
- M. Stredansky, A. Sala, T. Fontanot, R. Costantini, C. Africh, G. Comelli, L. Floreano, A. Morgante and A. Cossaro, Chem. Commun., 2018, 54, 3971–3973 RSC.
- D. Toffoli, M. Stredansky, Z. Feng, G. Balducci, S. Furlan, M. Stener, H. Ustunel, D. Cvetko, G. Kladnik, A. Morgante, A. Verdini, C. Dri, G. Comelli, G. Fronzoni and A. Cossaro, Chem. Sci., 2017, 8, 3789–3798 RSC.
- M. Kikuchi, T. Yamagishi and M. Hida, Dyes Pigm., 1981, 2, 143–151 CrossRef CAS.
- J. O. Morley, J. Phys. Chem., 1995, 99, 5956–5960 CrossRef CAS.
- G. Pawin, K. L. Wong, K.-Y. Kwon and L. Bartels, Science, 2006, 313, 961–962 CrossRef CAS PubMed.
- T. Kumagai, F. Hanke, S. Gawinkowski, J. Sharp, K. Kotsis, J. Waluk, M. Persson and L. Grill, Phys. Rev. Lett., 2013, 111, 246101 CrossRef PubMed.
- T. Kumagai, F. Hanke, S. Gawinkowski, J. Sharp, K. Kotsis, J. Waluk, M. Persson and L. Grill, Nat. Chem., 2014, 6, 41–46 CrossRef CAS PubMed.
- M. Koch, M. Pagan, M. Persson, S. Gawinkowski, J. Waluk and T. Kumagai, J. Am. Chem. Soc., 2017, 139, 12681–12687 CrossRef CAS.
- L. Floreano, A. Cossaro, R. Gotter, A. Verdini, G. Bavdek, F. Evangelista, A. Ruocco, A. Morgante and D. Cvetko, J. Phys. Chem. C, 2008, 112, 10794–10802 CrossRef CAS.
- R. Costantini, M. Stredansky, D. Cvetko, G. Kladnik, A. Verdini, P. Sigalotti, F. Cilento, F. Salvador, A. De Luisa, D. Benedetti, L. Floreano, A. Morgante, A. Cossaro and M. Dell’Angela, J. Electron Spectrosc. Relat. Phenom., 2018, 229, 7–12 CrossRef CAS.
- M. Klues, K. Hermann and G. Witte, J. Chem. Phys., 2014, 140, 14302 CrossRef PubMed.
- A. Cossaro, M. Puppin, D. Cvetko, G. Kladnik, A. Verdini, M. Coreno, M. De Simone, L. Floreano and A. Morgante, J. Phys. Chem. Lett., 2011, 2, 3124–3129 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental and theoretical details, NEXAFS spectra, additional STM images, and DFT calculation. See DOI: 10.1039/c9cc09915h |
| This journal is © The Royal Society of Chemistry 2020 |