
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Benchtop access to anhydrous actinide N-donor coordination complexes using ionic liquids†
Steven P.
Kelley
 a,
Volodymyr
Smetana
b,
Stephen D.
Emerson
c,
Anja-Verena
Mudring
*b and
Robin D.
Rogers
*bc
a,
Volodymyr
Smetana
b,
Stephen D.
Emerson
c,
Anja-Verena
Mudring
*b and
Robin D.
Rogers
*bc
aDepartment of Chemistry, University of Missouri, Columbia, MO 65211, USA
bDepartment of Materials and Environmental Chemistry, Stockholm University, 106 91 Stockholm, Sweden. E-mail: anja-verena.mudring@mmk.su.se
cCollege of Arts & Sciences, The University of Alabama, Tuscaloosa, AL 35487, USA. E-mail: rdrogers@ua.edu
First published on 10th March 2020
Abstract
By dehydrating actinide salts with an ionic liquid containing a common anion and subsequent reaction with N-heterocyclic ligands, we challenge the concept that actinides prefer O- over N-donors; rather the acidic hydrogen atoms of protic solvents hinder the formation of more elusive f-element N-donor coordination complexes.
As accumulating spent nuclear fuel poses a greater and greater hazard,1 the need to understand phenomena such as actinide–lanthanide separations2 or actinide speciation in the environment3 is becoming more critical. These in turn require an understanding of the basic coordination chemistry of actinide ions, and the nature of interactions between actinides and moderately soft donors (such as nitrogen) is an important, ongoing question as soft donors are commonly used as actinide-selective ligands in actinide–lanthanide separations.4
As N-heterocycles are found in the binding sites of promising new actinide-selective ligands5 and in proteins such as transferrin which are responsible for actinide transport in organisms,6 their interactions with actinides are of particular interest. Such interactions have been heavily studied in aqueous solution through spectroscopy and partitioning studies, but crystallographic studies, which would provide considerably more detail, are limited. Crystalline complexes of actinides and N-heterocycles are difficult to obtain from aqueous systems as N-heterocycles are often readily protonated while actinides tend to hydrolyze in concentrated aqueous solutions.7 Hydrolysis can be avoided by using strongly basic counterions, but such bases form strong interactions with actinide ions which dominate the coordination chemistry.8
Non-aqueous systems are often used to isolate unusual or weakly bound metal complexes, but they usually require the preparation of reactive intermediates from more readily available hydrated metal starting materials.9 While this can certainly be done with actinides,10 for transuranic elements this approach is only viable in a few specialized research institutions. Academic laboratories which have prepared crystalline transuranic compounds for structure determination have had to develop their own systems which allow isolation of new products from stock solutions or reagents in as few steps as possible.11
In trying to develop simple routes to N-donor complexation of transuranic ions, we hypothesized that since ionic liquids (ILs) can dissolve metal salts and are somewhat thermally stable, they could be used to easily isolate anhydrous salts which would be more likely to coordinate normally noncoordinating species. To test this, we chose the two most abundant actinides, thorium-232 and uranium-238, in their most stable oxidation states, U(VI) and Th(IV), which are strongly acidic and prone to polymerization through hydrolysis.12 Thorium(IV) Lewis base adducts have been made from thorium tetrahalides in strictly anhydrous conditions.13 Weakly ligated uranyl ([UO2]2+) complexes such as triflate salts14 or UO2Cl2(THF)3,15 can be used to make Lewis base adducts, but these typically must be isolated as intermediates and purified. We sought instead to dehydrate the hydrated nitrate salts of these elements with a nitrate IL on the benchtop, and react the resulting dehydrated compounds or solutions with N-heterocyclic ligands, again on the benchtop with no special precautions to exclude the surrounding atmosphere (Scheme 1).
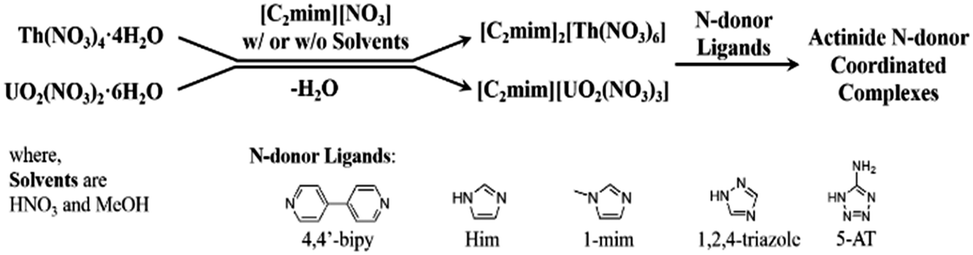 | ||
| Scheme 1 Dehydration and complexation strategy. | ||
We started by dissolving UO2(NO3)2·6H2O and Th(NO3)4·4H2O in 1-ethyl-3-methylimidazolium nitrate ([C2mim][NO3]) with and without solvent. Volatilization of the water and solvents (if present) led to the anhydrous salts [C2mim][UO2(NO3)3] as a supercooled liquid and [C2mim]2[Th(NO3)6] as a crystalline solid which were obtained quantitatively and in large scale. Subsequent reactions of these dehydrated starting materials with imidazole (H-Im), 1-methylimidazole (1-mim), 5-aminotetrazole (5-AT), 1,2,4-triazole (1,2,4-Triaz), and 4,4′-bipyridyl (4,4′-bipy) led easily to several N-donor complexes.
We employed two different strategies to prepare anhydrous Th(IV) nitrate salts. In the first method (see ESI† for experimental details), methanolic solutions of Th(NO3)4·4H2O and [C2mim][NO3] (a crystalline solid at RT, mp = 40 °C16) were combined in 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 molar ratio, resulting in the formation of crystalline [C2mim]2[Th(NO3)6] within minutes. After crystallization was complete, additional crystallization was observed when more [C2mim][NO3] was added to the solution. In this fashion, [C2mim]2[Th(NO3)6] was prepared on gram scale and isolated by filtration and drying to give 85% yield. The isolated crystals could be dried by heating for 2 h at 70 °C under ambient pressure and showed no apparent tendency to absorb moisture upon handling in air. IR spectroscopy indicates the material has a very sub-stoichiometric amount of water or methanol perhaps adsorbed to the surface (Fig. S2, ESI†). These crystals were used in subsequent coordination studies.
:2 molar ratio, resulting in the formation of crystalline [C2mim]2[Th(NO3)6] within minutes. After crystallization was complete, additional crystallization was observed when more [C2mim][NO3] was added to the solution. In this fashion, [C2mim]2[Th(NO3)6] was prepared on gram scale and isolated by filtration and drying to give 85% yield. The isolated crystals could be dried by heating for 2 h at 70 °C under ambient pressure and showed no apparent tendency to absorb moisture upon handling in air. IR spectroscopy indicates the material has a very sub-stoichiometric amount of water or methanol perhaps adsorbed to the surface (Fig. S2, ESI†). These crystals were used in subsequent coordination studies.
In the second dehydration method, [C2mim]2[Th(NO3)6], could be prepared quantitatively in gram scale without extra solvents by heating Th(NO3)4·4H2O with 2 eq. of [C2mim][NO3] at 90 °C overnight to remove water. The resulting product contained well formed crystals of a different polymorph of [C2mim]2[Th(NO3)6] residing in a liquid. The simplicity of this approach suggests an easy way to isolate anhydrous f-element salts amenable to the study of transuranic elements.
The uranyl salt, UO2(NO3)2·6H2O is known to hydrolyze in solutions containing dialkylimidazolium ILs to give salts of the [UO2(OH)2(NO3)2]2− anion,17 although Bradley et al. have reported obtaining an anhydrous 1,3-dimethylimidazolium salt of [UO2(NO3)4]2− from nitric acid solutions.18 In our approach, we dissolved UO2(NO3)2·6H2O with 1, 2, or 3 mol eq. of [C2mim][NO3] as a 50 wt% solution in concentrated nitric acid and evaporated the solutions for 5–6 days at 90 °C to give a non-volatile yellow liquid (see ESI†). Crystals formed in the reactions containing 2 or 3 mol eq. of IL after 5–6 days of standing at room temperature, one of which was analyzed by SCXRD and identified as [C2mim][UO2(NO3)3] (Fig. S1, right, ESI†). This complex has never been crystallized from an IL, but is known to be a major species in solution in ILs.19
The reaction time and presence of excess IL make it impractical to isolate [C2mim][UO2(NO3)3] in bulk by crystallization from nitric acid, and we thus investigated the solventless method shown above to work for Th(IV). Heating equimolar mixtures of UO2(NO3)2·6H2O and [C2mim][NO3] and at 70 °C under a current of N2 or dry air for at least 2 h produced yellow liquids with water contents below the threshold of detection by FTIR spectroscopy (Fig. S3, ESI†). The yellow liquids did not produce any crystals on cooling to room temperature; however, the IR spectrum contains two very strong peaks at 1514 and 1253 cm−1 which are consistent with the N–O stretching modes of bidentate nitrate ions similar to those in [C2mim][UO2(NO3)3].
The 1:1 stoichiometry of the reaction and the exclusively bidentate [NO3]− coordination suggested by the IR spectra indicate the yellow liquid to be supercooled [C2mim][UO2(NO3)3]. The fact that the liquid does not crystallize when prepared this way is not unprecedented;20 dialkylimidazolium halides (notably 1-butyl-3-methylimidazolium chloride ([C4mim]Cl)) are also known to form stable, supercooled liquids when they are especially dry.21 Thus, the solventless route affords essentially 100% yields of a pure supercooled [C2mim][UO2(NO3)3] liquid which is much easier to subsequently mix with our N-donors.
As part of our strategy to force coordination of N-donors, we sought similar solvents which would also dissolve the anhydrous salts, such as 1-methylimidazole, thus increasing the number of potential N-donor ligands. Reactions of [C2mim]2[Th(NO3)6] and 1,2,4-triazole in varying amounts of 1-mim at ratios of 1:1:n (n = 1–8) were conducted. Powder X-ray diffraction revealed the formation of a new crystalline phase at molar ratios of Th:1-mim greater than 2 (Fig. S4, ESI†). Single crystals of the compound were obtained by separately dissolving [C2mim]2[Th(NO3)6] and 1,2,4-triazole and combining the solutions. Colorless crystals formed from the resulting solution upon standing for 4–6 days at room temperature and were found by SCXRD to match the product phase (Fig. S5, ESI†). We had anticipated that the acidity of 1,2,4-triazole would result in an acid–base reaction with 1-mim to give an azolium azolate salt, which would exchange deprotonated triazolate anions with coordinated nitrate anions. However, the crystal structure of the product showed the formation of [H(1-mim)2][Th(NO3)5(1-mim)2] (Fig. 1 and Table S1, ESI†), which indicates that [C2mim]+ and [NO3]− are dissolved in the system, while excess 1-mim is able to coordinate to the metal ion and stabilize the [H(1-mim)]+ cation formed by deprotonation of the triazolate. Complexes of [Th(NO3)5L2]− anions with the same geometry are known for neutral O-donor ligands such as water,22 however, there are only two Th-imidazole and one Th-tetrazole crystallographically characterized coordination complexes reported to date.23
 | ||
| Fig. 1 Preparation of [H(1-mim)2][Th(NO3)5(1-mim)2] from [C2mim]2[Th(NO3)6]. The product is represented as a 50% probability ellipsoid plot of the formula unit with dashed lines indicating the shortest contacts between ions. | ||
Since [C2mim][UO2(NO3)3] could be isolated as either a crystalline solid or a supercooled liquid, both solventless and solution-based reactions were potentially available. Stoichiometric amounts of 1,2,4-Triaz, 5-AT, H-Im, 1-mim, and 4,4′-bipy were observed to either dissolve in hot [C2mim][UO2(NO3)3] or react with it to give solids after cooling (see ESI†). For the reaction with 4,4′-bipy, orange single crystals obtained from the mixture were characterized by SCXRD to be [C2mim]2[(UO2(NO3)3)2(4,4′-bipy)]·2{UO2(NO3)2(4,4′-bipy)} (Fig. 2 right). This cocrystal contains both anionic, dinuclear complexes and neutral, infinite coordination polymers where [UO2]2+ centers are bridged by 4,4′-bipy ligands. The neutral polymers and anionic molecules represent different levels of substitution of [NO3]− by 4,4′-bipy that would be expected to coexist in solution, and their co-crystallization likely occurs as a result of the neutral polymer buffering the extreme size difference between [(UO2(NO3)3)2(4,4′-bipy)]2− and [C2mim]+. We have observed similar, complicated crystalline phases to form when neutral co-formers are added to an IL with potentially strong intermolecular interactions.24
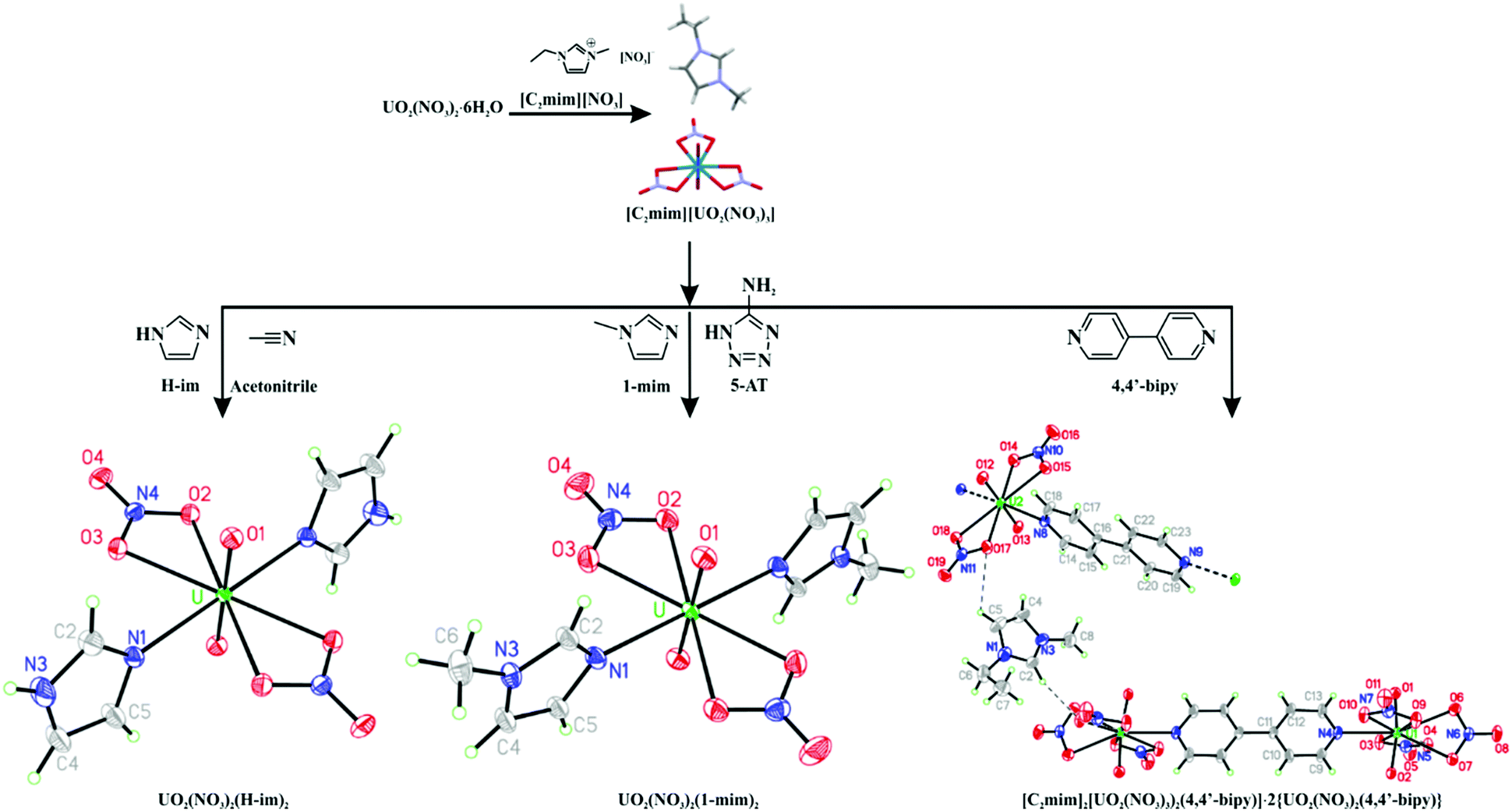 | ||
| Fig. 2 Preparation of UO2(NO3)2(H-Im)2 (left), UO2(NO3)2(1-mim)2 (middle), and [C2mim]2[(UO2(NO3)3)2(4,4′-bipy)]·2{UO2(NO3)2(4,4′-bipy)} (right) from [C2mim][UO2(NO3)3]. The products are represented as 50% probability ellipsoid plots of the formula units with dashed lines indicating the shortest contacts between ions and heavy dashes indicate extension of coordination polymer. | ||
The anionic [(UO2(NO3)3)2(4,4′-bipy)]2− has an extremely uncommon 9-coordinate uranyl center in which one [NO3]− is twisted out of the equatorial plane in a conformation that certainly weakens the metal–ligand interaction. It is interesting to note that this binding mode has only been observed for uranyl nitrate complexes, and many of these involve coordination to an N-donor ligand.25 The increase in bond distances may indicate a reduced positive charge on the metal centers (which would both weaken the metal–[NO3]+ interactions and increase the metal ionic radii) (see ESI†). This indicates that despite the lack of a negative charge, the N-heterocycles are better electron N-donors than [NO3]− which is consistent with the fact that they are stronger bases.
The reaction of 1 eq. of 5-AT with 2 eq. of 1-mim to form a putative IL produced a homogeneous solution after heating with a heat gun. Adding this solution to hot [C2mim][UO2(NO3)3] and heating at 110–120 °C overnight, resulted in a mixture of orange solids and gel from which single crystals of UO2(NO3)2(1-mim)2 (Fig. 2 middle) could be isolated. UO2(NO3)2(1-mim)2 has been detected in solution26 and predicted in the gas phase,27 but has never been isolated as a crystalline solid, probably due to hydrolysis at higher concentrations. It is practically isostructural to the reported UO2(OAc)2(1-mim)2 (OAc = acetate anion).27
Finally, we investigated the use of a solvent to control the crystallization. Two eq. of H-Im and supercooled [C2mim][UO2(NO3)3] were dissolved in ∼1 mL of dry acetonitrile. The resulting mixture was evaporated in a Drierite-filled desiccator at reduced pressure to produce large yellow crystals of UO2(NO3)2(H-Im)2 (Fig. 2 left) after 4 days. This compound exhibits U coordination identical to UO2(NO3)2(1-mim)2 and UO2(OAc)2(1-mim)2, but crystallizes in the monoclinic space group P21/c (Table S1, ESI†).
The N-heterocyclic ligands appear to have a significant effect on metal–[NO3]− interactions. It is known that actinide metal–ligand bonds are correlated with coordination number, but in all cases where the coordination number remained unchanged, coordination of the N-heterocyclic ligand resulted in a significant increase of all metal–[NO3]− bond distances (Table S2, ESI†). Besides geometric reasons this may serve as a proof of enhanced metal–N-donor interactions. On the other hand, N-heterocyclic ligands are also known to have significant influence on the crystal packing not at least due to non-covalent interactions.28
The protonation of H-Im and 4,4′-bipy by UO2(NO3)2·6H2O to give ammonium salts is established in the literature.29 The structures here are all analogous to UO2(NO3)2(OH2)230 except for the [(UO2(NO3)3)2(4,4′-bipy)]2− anion which has a 9-coordinate uranium center. Thus, while a number of reactions of UO2(NO3)2·6H2O with N-heterocycles (including 1-mim, H-Im, and 4,4′-bipy) have already been reported, the crystallization of uranyl nitrate complexes with these ligands is unprecedented. It is difficult to obtain the crystals of uranyl nitrate complexes with N-heterocycles from aqueous medium as N-heterocycles can readily be protonated and also the actinides tend to hydrolyze in concentrated aqueous solutions. Here, we successfully obtained the crystals of those novel complexes by simply reacting N-heterocycles with dehydrated liquid uranyl nitrate.
While complexes with N-heterocyclic ligands can be isolated in the presence of more basic anions, the use of more weakly binding [NO3]− counterions in the absence of water should lead to greater flexibility of the [UO2]2+ coordination geometry. This has been illustrated here by the fortuitous isolation of two distinct species in the crystal structure of [C2mim]2[(UO2(NO3)3)2(4,4′-bipy)]·2{UO2(NO3)2(4,4′-bipy)}. By comparison, six crystalline complexes of [UO2]2+ with N-heterocycles have also been crystallized from reactions with UO2(NO3)2·6H2O and acetylacetone in solution.31 However, this approach only resulted in the isolation of complexes with the same coordination geometries (two bidentate acetylacetonate ions and one unidentate N-donor), and prevented the isolation of any complexes with a bidentate N-heterocycle (2,2′-bipyridyl).
In summary, a suitably simple approach for the isolation of transuranic N-donor complexes was developed and illustrated with the ready dehydration of Th(NO3)4·4H2O and UO2(NO3)2·6H2O in an IL followed by complexation with aromatic N-donor ligands thus eliminating the acidic hydrogen atoms from water or other protic solvents which compete with the f-elements for access to the N-donor and affording readily crystallizable complexes. The isolated compounds include [H(1-mim)2][Th(NO3)5(1-mim)2], only the fourth Th-azole coordination complex reported to date, [C2mim][UO2(NO3)3] which has never been crystallized from an IL but is known to be a major species in solution in ILs, and the unprecedented crystallization of uranyl nitrate complexes of 1-mim, H-Im, and 4,4′-bipy, including UO2(NO3)2(1-mim)2 which has been detected in solution and predicted in the gas phase, but never isolated as a crystalline solid. The synthetic approaches developed here, showed that despite the lack of a negative charge, the N-heterocycles are better electron donors than [NO3]− which is consistent with the fact that they are stronger bases. It is apparent that ILs will play a key role in the exploration of this chemistry by providing redox-inert, anhydrous environments, amenable to small scale benchtop manipulations for compounds which cannot normally be dissolved in organic solvents.
This research was supported in part through the Göran Gustafsson prize by the Royal Swedish Academy of Science to A.-V. M. We also thank the U.S. Department of Energy Basic Energy Sciences, Heavy Elements program under Award Number DE-SC0019220 and Vetenskapsrådet for a Tage Erlander Professorship (VR grant 2018-00233) for support.
Conflicts of interest
There are no conflicts to declare.Notes and references
- J. Johnson, Chem. Eng. News, 2014, 92, 22–23 Search PubMed.
- K. L. Nash, in Handbook on the Physics and Chemistry of Rare Earths, ed. K. A. Gschneider Jr., L. Eyring, G. R. Choppin and G. H. Lander, Elsevier Science, Amesterdam, 1994, vol. 18, pp. 197–238 Search PubMed.
- G. R. Choppin, Radiochim. Acta, 2003, 91, 645–650 CAS.
- (a) M. Nilsson and K. L. Nash, Solvent Extr. Ion Exch., 2007, 25, 665–701 CrossRef CAS; (b) M. J. Hudson, L. M. Harwood, D. M. Laventine and F. W. Lewis, Inorg. Chem., 2013, 52, 3414–3428 CrossRef CAS PubMed.
- F. W. Lewis, L. M. Harwood, M. J. Hudson, M. G. B. Drew, J. F. Desreux, G. Vidick, N. Bouslimani, G. Modolo, A. Wilden, M. Sypula, T.-H. Vu and J.-P. Simonin, J. Am. Chem. Soc., 2011, 133, 13093–13102 CrossRef CAS PubMed.
- J. B. Vincent and S. Love, Biochim. Biophys. Acta, Gen. Subj., 2012, 1820, 362–378 CrossRef CAS PubMed.
- Z. Kolarik, Chem. Rev., 2008, 108, 4208–4252 CrossRef CAS PubMed.
- G. R. Choppin and M. P. Jensen, in The Chemistry of the Actinide and Transactinide Elements, ed. L. R. Morss, N. M. Edelstein and J. Fuger, Springer, Netherlands, Dordrecht, 2006, pp. 2524–2621 Search PubMed.
- (a) A. Babai and A.-V. Mudring, Dalton Trans., 2006, 1828–1830 RSC; (b) A. Babai and A.-V. Mudring, Inorg. Chem., 2006, 45, 3249–3255 CrossRef CAS PubMed; (c) S.-F. Tang and A.-V. Mudring, Cryst. Growth Des., 2009, 9, 2549–2551 CrossRef CAS; (d) S. Tang and A.-V. Mudring, Cryst. Growth Des., 2011, 11, 1437–1440 CrossRef CAS.
- (a) S. D. Reilly, J. L. Brown, B. L. Scott and A. J. Gaunt, Dalton Trans., 2014, 43, 1498–1501 RSC; (b) A. J. Gaunt, A. E. Enriquez, S. D. Reilly, B. L. Scott and M. P. Neu, Inorg. Chem., 2008, 47, 26–28 CrossRef CAS PubMed; (c) A. J. Gaunt, S. D. Reilly, T. W. Hayton, B. L. Scott and M. P. Neu, Chem. Commun., 2007, 1659–1661 RSC.
- (a) R. E. Wilson, D. D. Schnaars, M. B. Andrews and C. L. Cahill, Inorg. Chem., 2014, 53, 383–392 CrossRef CAS PubMed; (b) G. B. Jin, Y.-J. Hu, B. Bellott, S. Skanthakumar, R. G. Haire, L. Soderholm and J. A. Ibers, Inorg. Chem., 2013, 52, 9111–9118 CrossRef CAS PubMed.
- G. R. Choppin, Mar. Chem., 2006, 99, 83–92 CrossRef CAS.
- M. G. B. Drew and G. R. Willey, J. Chem. Soc., Dalton Trans., 1984, 727–729 RSC.
- Jean C. Berthet, M. Lance, M. Nierlich and M. Ephritikhine, Eur. J. Inorg. Chem., 2000, 1969–1973 CrossRef CAS.
- M. P. Wilkerson, C. J. Burns, R. T. Paine and B. L. Scott, Inorg. Chem., 1999, 38, 4156–4158 CrossRef CAS.
- S. Zhang, X. Lu, Q. Zhou, X. Li, X. Zhang and S. Li, Ionic liquids: physicochemical properties, Elsevier, 2009 Search PubMed.
- (a) S. P. Kelley, J. S. Nuss and R. D. Rogers, Application of Ionic Liquids on Rare Earth Green Separation and Utilization, Springer, 2016, pp. 21–42 Search PubMed; (b) F. Qu, Q.-Q. Zhu and C.-L. Liu, Cryst. Growth Des., 2014, 14, 6421–6432 CrossRef CAS.
- A. E. Bradley, C. Hardacre, M. Nieuwenhuyzen, W. R. Pitner, D. Sanders, K. R. Seddon and R. C. Thied, Inorg. Chem., 2004, 43, 2503–2514 CrossRef CAS PubMed.
- K. Servaes, C. Hennig, I. Billard, C. Gaillard, K. Binnemans, C. Görller-Walrand and R. Van Deun, Eur. J. Inorg. Chem., 2007, 5120–5126 CrossRef CAS.
- A.-V. Mudring, Aust. J. Chem., 2010, 63, 544–564 CrossRef CAS.
- C. P. Fredlake, J. M. Crosthwaite, D. G. Hert, S. N. V. K. Aki and J. F. Brennecke, J. Chem. Eng. Data, 2004, 49, 954–964 CrossRef CAS.
- G. E. Sigmon and P. C. Burns, J. Solid State Chem., 2010, 183, 1604–1608 CrossRef CAS.
- (a) C. Zhang, G. Hou, G. Zi and M. D. Walter, Dalton Trans., 2019, 48, 2377–2387 RSC; (b) C. Zhang, G. Hou, G. Zi, W. Ding and M. D. Walter, Inorg. Chem., 2019, 58, 1571–1590 CrossRef CAS PubMed; (c) K. P. Browne, K. A. Maerzke, N. E. Travia, D. E. Morris, B. L. Scott, N. J. Henson, P. Yang, J. L. Kiplinger and J. M. Veauthier, Inorg. Chem., 2016, 55, 4941–4950 CrossRef CAS PubMed.
- H. M. Titi, S. P. Kelley, M. E. Easton, S. D. Emerson and R. D. Rogers, Chem. Commun., 2017, 53, 8569–8572 RSC.
- J.-C. Berthet, P. Thuéry, J.-P. Dognon, D. Guillaneux and M. Ephritikhine, Inorg. Chem., 2008, 47, 6850–6862 CrossRef CAS PubMed.
- A. Marzotto and H. Kozłowski, Inorg. Chim. Acta, 1982, 67, 87–90 CrossRef CAS.
- K. E. Gutowski, V. A. Cocalia, S. T. Griffin, N. J. Bridges, D. A. Dixon and R. D. Rogers, J. Am. Chem. Soc., 2007, 129, 526–536 CrossRef CAS PubMed.
- E. A. Meyer, R. K. Castellano and F. Diederich, Angew. Chem., Int. Ed., 2003, 42, 1210–1250 CrossRef CAS PubMed.
- (a) D. L. Perry, H. Ruben, D. H. Templeton and A. Zalkin, Inorg. Chem., 1980, 19, 1067–1069 CrossRef CAS; (b) N. W. Alcock and D. J. Flanders, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 1987, 43, 1267–1269 CrossRef.
- D. Hall, A. D. Rae and T. N. Waters, Acta Crystallogr., 1965, 19, 389–395 CrossRef CAS.
- T. Kawasaki, T. Nishimura and T. Kitazawa, Bull. Chem. Soc. Jpn., 2010, 83, 1528–1530 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Materials and methods, characterizations, crystallographic details. CCDC 1968006–1968012. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c9cc09852f |
| This journal is © The Royal Society of Chemistry 2020 |