
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceHydrogen release from liquid organic hydrogen carriers catalysed by platinum on rutile-anatase structured titania†
P. T.
Aakko-Saksa
 *ab,
M.
Vehkamäki
a,
M.
Kemell
a,
L.
Keskiväli
b,
P.
Simell
b,
M.
Reinikainen
b,
U.
Tapper
b and
T.
Repo
*a
*ab,
M.
Vehkamäki
a,
M.
Kemell
a,
L.
Keskiväli
b,
P.
Simell
b,
M.
Reinikainen
b,
U.
Tapper
b and
T.
Repo
*a
aUniversity of Helsinki, A. I. Virtasen aukio 1, PO Box 55, 00014 Helsinki, Finland. E-mail: timo.repo@helsinki.fi; paivi.aakko-saksa@helsinki.fi
bVTT Technical Research Centre of Finland Ltd, PO Box 1000, 02044 VTT, Finland. E-mail: paivi.aakko-saksa@vtt.fi
First published on 8th January 2020
Abstract
A liquid organic hydrogen carrier (LOHC) is an interesting concept for hydrogen storage. We describe herein a new, active catalyst system for dehydrogenation of perhydrogenated dibenzyl toluene (H18-DBT), which is a promising LOHC candidate. Pt supported on a rutile-anatase form of titania was found to be more active than Pt supported on anatase-only titania, or on alumina, and almost equally active as Pt supported on carbon. Robust and durable metal oxide supports are preferred for catalysing reactions at high temperatures.
Hydrogen is a flexible energy carrier. However, its storage is challenging. In this respect, LOHCs are promising as reversible, effective, safe, user-friendly and economic forms of hydrogen storage. An interesting LOHC pair is based on dibenzyl toluene (DBT) and its perhydrogenated counterpart (H18-DBT) with a reasonable hydrogen storage capacity of 6.2 wt%, and good compatibility with the existing fuel infrastructure owing to its liquid form and stability at normal temperature and pressure.1 Catalysts for dehydrogenation of LOHC candidates are not equal regarding their activity and durability.2–8 Choices of catalytic metal, support and preparation procedures are decisive for catalyst properties.9,10
The dehydrogenation of cycloalkanes is challenging since it is necessary to break the strong C–H bond. Furthermore, aromatics are abundant in crude oil, diminishing the need for their synthesis and the development of selective dehydrogenation catalysts for this purpose. However, non-selective reforming process is common in refining industries using, for example, noble metal catalysts, particularly those based on Pt.7,11–13 Brückner et al.14 found Pt on carbon to be the most active catalyst amongst those studied, and Pt on alumina (Al2O3) the second most active, while Pt/SiO2, Pd/C and Pd/Al2O3 were not substantially active for the dehydrogenation of H18-DBT. The conversion of H18-DBT was over 95% using Pt/Al2O3 catalyst at 290 °C in 3.5 hours, and faster hydrogen release at 310 °C was reported.14 A hydrogen release of 50% was reported by Fikrt et al.15 for a Pt/Al2O3 catalyst at 0.5 wt% metal loading for hydrogen release in continuous operation at a temperature range from 290 to 320 °C. Carbon support is cost effective and abundant. However, more durable metal oxide supports are desired for dehydrogenation of H18-DBT as the reaction takes place at a high temperature, close to 300 °C. Additionally, metal oxide supports are highly active for hydrogen spillover, while for carbon supports this property is compensated to some extent by chemisorption. Proper hydrogen spillover viz. migration of hydrogen over support16 is needed besides breaking hydrogen carbon bond as the dehydrogenation catalyst should desorb hydrogen. For challenging dehydrogenation of cycloalkanes, metal oxide supports are more practical than carbon supports.
Interestingly, higher activity has been observed for catalysts supported on titania and lanthanum oxide than on alumina for the dehydrogenation of methylcyclohexane (MCH).7,17 Additionally, notable are general features of TiO2 nanopowder supports, including strong metal–support interaction, chemical stability, acid–base property and electrocatalytic properties. Titania nanoparticles are commonly used in water-splitting reaction,18,19 and have been suggested as an alternative to Pt/C in PEM fuel cells.20 These observations inspired us to study Pt on titania supports for hydrogen release from H18-DBT. We prepared Pt catalysts supported on titania formed of rutile and anatase (Pt/TiO2ra) for the dehydrogenation of H18-DBT (Fig. 1). The activity of this catalyst was compared with the Pt supported on titania formed of anatase only (Pt/TiO2ana), and on alumina (Pt/γ-Al2O3). We present findings with in-house catalysts (two titania and one alumina) in comparison with a commercial Pt/C catalyst for hydrogen release from H18-DBT substrate.
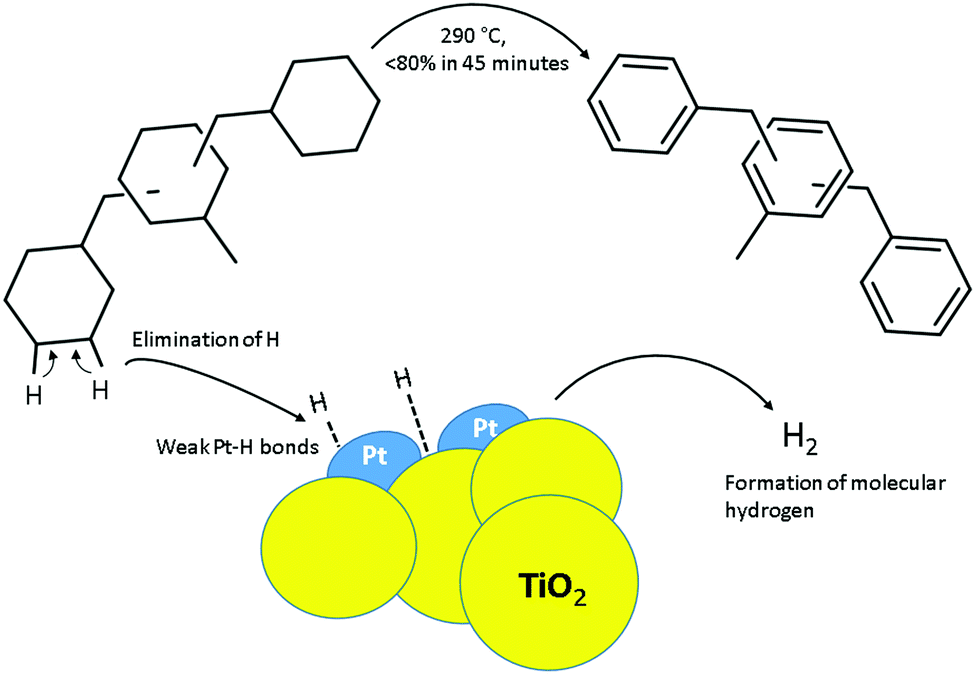 | ||
| Fig. 1 Schematic presentation of hydrogen release from H18-DBT using Pt on titania catalyst. | ||
Catalysts were prepared by incipient wetness impregnation using chloroplatinic acid (H2PtCl6) with a target Pt concentration of 1 wt%. The catalysts were calcinated at 500 °C in air for two hours, which did not induce significant phase transition. Pt/TiO2ra contained 86.1 wt% anatase and Pt/TiO2ana 100% anatase after calcination (Table 1, details in ESI†). Three catalyst types were prepared in multiple batches to consider the repeatability of preparation. The hydrogen release rate from H18-DBT substrate with 95% hydrogenation degree was evaluated at 290 °C. A mixture of H18-DBT and the catalyst were heated in a round bottom flask while stirring with a magnetic stirrer at 300 min−1. The molar ratio of the substrate to Pt was 400![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1. Dehydrogenation degree was quantitatively analysed by NMR.21,22 The results are shown as a percentage of the hydrogen released of a theoretical maximum in 45 minutes at 290 °C (Table 1 and Fig. 2, details in ESI†) and as cumulatively in selected tests (Fig. 3).
:1. Dehydrogenation degree was quantitatively analysed by NMR.21,22 The results are shown as a percentage of the hydrogen released of a theoretical maximum in 45 minutes at 290 °C (Table 1 and Fig. 2, details in ESI†) and as cumulatively in selected tests (Fig. 3).
| Pt/γ-Al2O3 | Pt/TiO2ra | Pt/TiO2ana | |
|---|---|---|---|
| a Substrate H18-DBT, temperature 290 °C. b Manufacturer's data. c Results in ESI for ICP, S/TEM, EDS, XRD, XPS, NMR, H2 release. d Several catalyst batches prepared (details in ESI). | |||
| Structure | Gamma 100%b | Anatase 86% | Anatase 100%c |
| Rutile 14%c | |||
| Pt content (wt%) | 0.9 ± 0.1c | 0.9 ± 0.1c | 0.9 ± 0.1c |
| Particle size of support (μm) | 40b | 0.025b | 0.020b |
| Particle size, Pt (nm) | <1c | 1.3c | |
| Hydrogen release, 45 min. (% th. max.) | 55 ± 4a,c | 65 ± 5a,c | 32 ± 5a,c |
| Replicates | n = 6 (2 cat.c,d) | n = 11 (5 cat.c,d) | n = 5 (3 cat.c,d) |
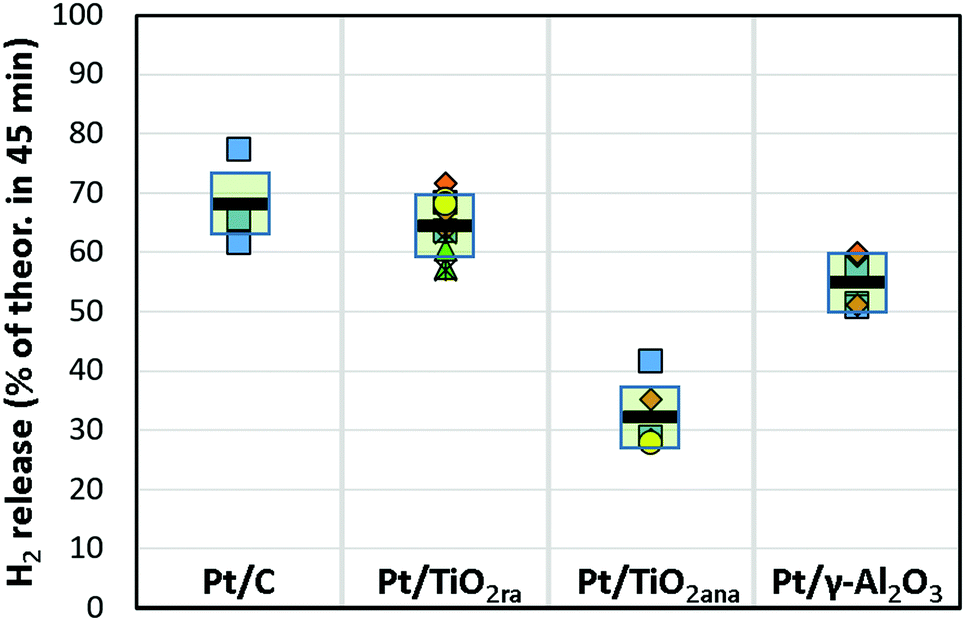 | ||
| Fig. 2 Hydrogen release from H18-DBT using Pt (1 wt%) catalysts on carbon, titania and alumina supports as a percentage of the theoretical maximum in 45 minutes. A temperature was 290 °C. A molar ratio of substrate to Pt was 400:1. Markers represent different catalyst batches and horizontal lines are the average results. | ||
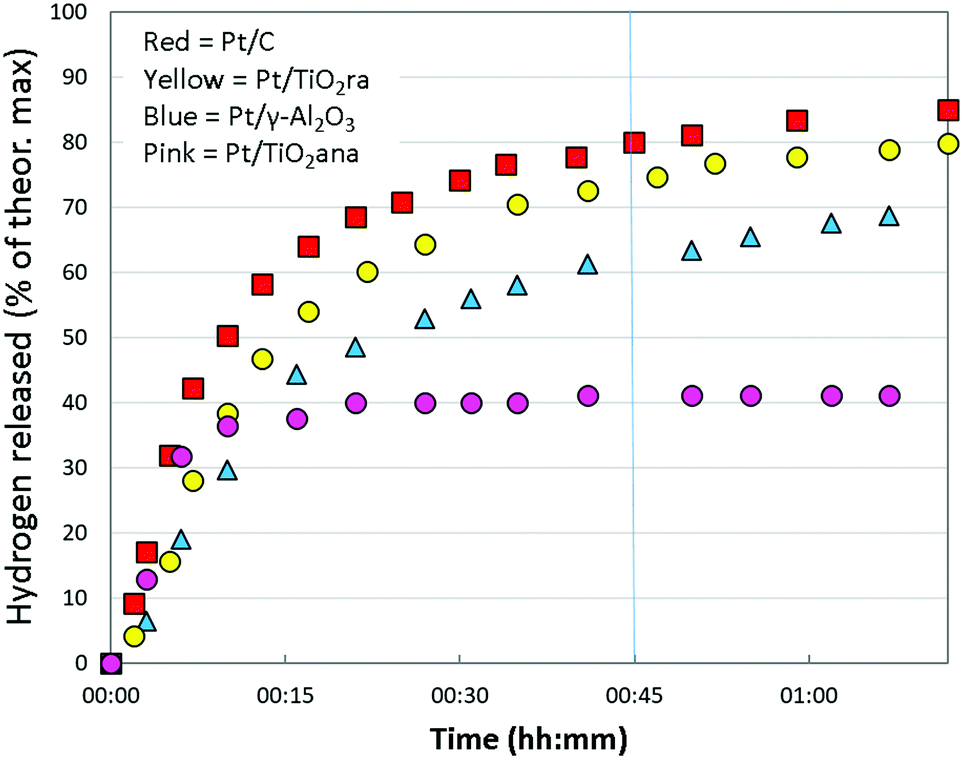 | ||
| Fig. 3 Hydrogen release from H18-DBT using Pt (1 wt%) supported on carbon, titania and alumina in the selected tests. T = 290 °C. | ||
In the series, the Pt catalyst supported on titania nanopowder with both anatase and rutile forms was substantially active, showing a hydrogen release rate of approx. 65% in 45 minutes at 290 °C. This was close to the activity of the Pt/C catalyst that was capable of releasing 68% of hydrogen (on average) in the same conditions. Instead, activity of the Pt catalyst prepared on titania nanopowder with anatase only form, a common catalyst support, was only modest showing on average 32% hydrogen release of the theoretical maximum. We further extended our studies to Pt on a high surface area pore-controlled γ-Al2O3, which proved to be active, but not to the extent of a Pt/TiO2ra. Pt/γ-Al2O3 enabled hydrogen release of approx. 55% of the theoretical maximum in 45 minutes from H18-DBT. Turnover frequency was on average 3420 h−1 for Pt/TiO2ra (details in ESI†). In our work, the most active catalyst among those studied was the new concept of Pt on TiO2 with both anatase and rutile forms, showing almost as high activity as the Pt/C catalyst used as a reference. To our knowledge, the capability of TiO2ra to support highly active Pt for dehydrogenation of cyclohexyl structures is unpresented.
Several batches of in-house catalysts were prepared and evaluated in replicates in order to study the consistency of the results (Fig. 2), which depend on the Pt concentrations of catalysts, molar ratios of substrate to Pt and control of reaction temperature. Pt concentrations of the catalysts (0.9 ± 0.1 wt%) were deemed sufficiently narrow range for comparisons of different catalyst types. The uncertainty of the molar ratio of substrate to Pt (400:1) relates to the uncertainty of Pt concentrations. The reaction temperature controlled by a hot plate varied slightly. Even when considering the uncertainty of the measurements, higher activities of Pt/TiO2ra and Pt/C catalysts than of Pt/γ-Al2O3 and Pt/TiO2ana catalysts studied appear to be significant for the dehydrogenation of H18-DBT.
At the beginning of the test, the dehydrogenation degree from H18-DBT was almost equal for different catalysts (Fig. 3). In higher conversions, the reaction proceeded faster with the Pt/C and Pt/TiO2ra catalysts than with the other catalysts. When the reaction reached a 70% dehydrogenation degree, Pt/TiO2ra was almost as efficient as Pt/C. Dehydrogenation with a Pt/γ-Al2O3 catalyst was relatively fast, although not to the same extent as with Pt/TiO2ra or Pt/C catalysts. Unexpectedly, dehydrogenation with Pt/TiO2ana ceased and the catalyst typically became completely inactive. Complete dehydrogenation of H18-DBT was not achieved in the given time (1 h 15 min) with any of the catalysts. Based on the above, Pt/TiO2ra was deemed to enable fast and smooth hydrogen release from H18-DBT.
The high activity of the Pt/TiO2ra catalyst for dehydrogenation of H18-DBT may relate to the properties of Pt particles, the support or their interactions with each other, or with the substrate. A high surface area of a support is generally favourable for catalysis. However, this did not seem to explain the high activity of the Pt/TiO2ra catalyst in our experiments. The surface area of TiO2 supported catalysts was lower (below 80 m2 g−1)18,19 than the surface area of γ-Al2O3 support (100 m2 g−1), particularly when compared to carbon supports that typically have high surface areas (200–300 m2 g−1).20 Nanosized particles of the support might be efficient in catalysis. However, in these experiments, the catalyst supported on the TiO2ra nanoparticles showed a high degree of dehydrogenation, while the other catalyst supported on TiO2ana nanoparticles deactivated soon. TEM analyses confirmed a uniform particle size of TiO2ra particles (from 16 nm to 20 nm) and also of TiO2ana particles (from 12 nm to 26 nm) (Fig. 4, details in ESI†). These titania nanopowders had an agglomerated structure, although not sintered. The particle size of the alumina support was approx. 40 μm with a fine structure of pores in a size class of 10 nm. Overall, the catalyst activity in our work did not follow the order of the surface areas or particle sizes of the supports, while other physical properties of catalysts may be important.
 | ||
| Fig. 4 Images of Pt/TiO2ra by (a) TEM (b) high-angle annular dark-field STEM (images of Pt/TiO2ana, Pt/γ-Al2O3 and more details in ESI†). | ||
We further studied Pt particles of the titania nanopowder supported catalysts. For example, good dispersion of Pt is generally favourable for carbon supported catalysts.20 Pt particles on titania nanopowders Pt/TiO2ra and Pt/TiO2ana were extremely small: in the size class of 1–2 nm or even below 1 nm (Fig. 4, details in ESI†). Pt particles smaller than 2 nm have also previously been detected on the surface of TiO2 by Filippo et al.23 Sun et al.24 observed that the crystalline structure of Pt particles below 1 nm spontaneously collapses and has an amorphous nature. These Pt particles have only 32 atoms, and their molecular orbitals are relatively separated and localized, leading to a short Pt–Pt distance and a strong bond between Pt and adsorbed hydrogen. Shukla16 concluded that strong metal–support interaction rather than the particle size of Pt, or the ability of the support to keep Pt in a reduced state, explains the high activity of Pt on titania catalyst for dehydrogenation of MCH. The XPS results showed higher oxidation state for a part of Pt in Pt on TiO2ra than on TiO2ana (details in ESI†). Small Pt nanoparticles enhance formation of oxides, particularly when size of Pt particles is below 1.3 nm.25,26 High Lewis acidity of Pt particles on titania supports, and higher acidity of Pt on TiO2ra than on TiO2ana catalyst in combination with small size of Pt particles may partially explain the high activity of Pt/TiO2ra catalyst for hydrogen release from H18-DBT.
In our experiments, two titania-supported catalysts with Pt nanoparticles showed high activity for hydrogen release from H18-DBT, although one of these catalysts, TiO2ana, deactivated soon. These catalysts had seemingly similar physical properties, both were nanopowders with similar surface areas and Pt particles were also small (<2 nm). Only structural forms of supports were different, and oxidation state of Pt/TiO2ra was slightly higher than that of Pt/TiO2ana. Deactivation of catalysts can be many-fold, for example related to the agglomeration of the Pt particles and adsorption of polyaromatic molecules on the active sites of catalyst surface (analogous to coke adsorption).27,28 These adverse effects can be suppressed by high metal–support interaction, which stabilises small Pt particles.29 TiO2ra is characterised by rutile phase defect sites capable to bind Pt tightly30 and Pt(0) atoms are found to thermally diffuse into TiO2 lattice under oxidizing atmosphere31 leading to strong interaction between Pt and TiO2ra support. Furthermore, in Pt/TiO2ra catalyst, nanosized Pt particles were unevenly dispersed providing regions for migration of polyaromatic molecules from active metal sites. However, increasing share of rutile from 14% to 84% in TiO2 supported catalyst at 1 wt% Pt loading did not improve hydrogen release rate from H18-DBT in the preliminary tests. Indicatively, deactivation of Pt/TiO2ra catalyst was substantial for the second dehydrogenation cycle, which could be related to growth of Pt particles in used catalyst. Thus, small Pt particle size, strong metal support interaction and unevenly distributed Pt leaving regions for migration of polyaromatic molecules were thought to be important for Pt/TiO2ra catalyst, although, general properties of the titania support with both anatase and rutile forms are needed for Pt nanoparticles to sustain high catalytic activity and resistance towards deactivation.
In conclusion, hydrogen storage with LOHC is an interesting concept and we found an active catalyst system for dehydrogenation of such a candidate. Pt supported on a rutile-anatase form of titania nanopowder had high activity for dehydrogenation of H18-DBT at 290 °C, almost equally as active as Pt supported on a carbon catalyst that is known for its high level of activity. Metal oxide supports are robust and durable, and are therefore desirable for reactions at high temperatures rather than carbon supports. In our experiments, a Pt catalyst on alumina support was efficient, although not to the same extent as Pt supported on a rutile-anatase form of titania nanopowder, while Pt supported on anatase-only titania nanopowder deactivated in the early phase of reaction. Differences in the activities of the studied catalysts did not relate to the surface areas or particle sizes of the supports, while the characteristics of Pt particles, e.g. their size and Lewis acidity could be significant along with some support properties enhancing resistance of catalyst towards deactivation. Further research is ongoing in order to elucidate stability of the system and mechanisms behind the activity of the Pt catalyst supported on titania nanopowder with anatase and rutile forms for dehydrogenation of H18-DBT.
We gratefully acknowledge funding from Business Finland (No. 1508/31/2017) and industrial partners within the LOHCNESS project. We thank Mikko Heikkilä for XRD analyses, and the Electron Microscopy unit at the University of Helsinki for TEM instrument access.
Conflicts of interest
There are no conflicts of interest to declare.References
- K. Müller, K. Stark, V. N. Emelyanenko, M. A. Varfolomeev, D. H. Zaitsau, E. Shoifet, C. Schick, S. P. Verevkin and W. Arlt, Ind. Eng. Chem. Res., 2015, 54, 7967–7976 CrossRef
.
- P. Preuster, C. Papp and P. Wasserscheid, Acc. Chem. Res., 2017, 50, 74–85 CrossRef CAS PubMed
- Y. Okada, E. Sasaki, E. Watanabe, S. Hyodo and H. Nishijima, Int. J. Hydrogen Energy, 2006, 31, 1348–1356 CrossRef CAS
- R. H. Crabtree, Energy Environ. Sci., 2008, 134–138 RSC
-
M. J. Schneider, Bridging Renewable Electricity with Transportation Fuels, 2015 Search PubMed
- P. T. Aakko-Saksa, C. Cook, J. Kiviaho and T. Repo, J. Power Sources, 2018, 396, 803–823 CrossRef CAS
- A. Shukla, S. Karmakar and R. B. Biniwale, Int. J. Hydrogen Energy, 2011, 37, 3719–3726 CrossRef
- A. Bourane, M. Elanany, T. V. Pham and S. P. Katikaneni, Int. J. Hydrogen Energy, 2016, 41, 23075–23091 CrossRef CAS
- Y. Yu, O. Y. Gutiérrez, G. L. Haller, R. Colby, B. Kabius, J. A. R. Van Veen, A. Jentys and J. A. Lercher, J. Catal., 2013, 304, 135–148 CrossRef CAS
- M. Campanati, G. Fornasari and A. Vaccari, Catal. Today, 2003, 77, 299–314 CrossRef CAS
-
K. Owen and T. Coley, Automotive Fuels Reference Book, Society of Automotive Engineers, Warrendale, 1995 Search PubMed
- M. Taube and P. Taube, EIR-Ber., 1979, 54 Search PubMed
- C. N. M. Ouma, P. Modisha and D. Bessarabov, RSC Adv., 2018, 8, 31895–31904 RSC
- N. Brückner, K. Obesser, A. Bösmann, D. Teichmann, W. Arlt, J. Dungs and P. Wasserscheid, ChemSusChem, 2014, 7, 229–235 CrossRef PubMed
- A. Fikrt, R. Brehmer, V. Milella, K. Müller, A. Bösmann, P. Preuster, N. Alt, E. Schlücker, P. Wasserscheid and W. Arlt, Appl. Energy, 2017, 194, 1–8 CrossRef CAS
- A. A. Shukla, P. V. Gosavi, J. V. Pande, V. P. Kumar, K. V. R. Chary and R. B. Biniwale, Int. J. Hydrogen Energy, 2010, 35, 4020–4026 CrossRef CAS
- A. U. Pradhan, A. Shukla, J. V. Pande, S. Karmarkar and R. B. Biniwale, Int. J. Hydrogen Energy, 2010, 36, 680–688 CrossRef
- S. Bagheri, N. M. Julkapli and S. Bee Abd Hamid, Sci. World J., 2014, 1–22 Search PubMed
-
R. Julian, Heterogeneous Catalysis, Elsevier B.V., Amsterdam, 2012 Search PubMed
- J. Parrondo, T. Han, E. Niangar, C. Wang, N. Dale, K. Adjemian and V. Ramani, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 45–50 CrossRef CAS PubMed
- G. Do, P. Preuster, R. Aslam, A. Bosmann, K. Muller, W. Arlt and P. Wasserscheid, R. Soc. Chem., 2016, 1, 313–320 CAS
- L. Shi, S. Qi, J. Qu, T. Che, C. Yi and B. Yang, Int. J. Hydrogen Energy, 2019, 44, 5345–5354 CrossRef CAS
- E. Filippo, C. Carlucci, A. L. Capodilupo, P. Perulli, F. Conciauro, G. A. Corrente, G. Gigli and G. Ciccarella, Mater. Res., 2015, 18, 473–481 CrossRef CAS
- Y. Sun, L. Zhuang, J. Lu, X. Hong and P. Liu, JACS Commun., 2007, 15465–15467 CrossRef CAS PubMed
- C. Wang and C. Yeh, J. Catal., 1998, 178, 450–456 CrossRef CAS
- J. R. Croy, S. Mostafa, L. Hickman, H. Heinrich and B. R. Cuenya, Appl. Catal., A, 2008, 350, 207–216 CrossRef CAS
- H. N. Pham, J. J. H. B. Sattler, B. M. Weckhuysen and A. K. Datye, Catalysis, 2016, 2257–2264 CAS
- E. Gianotti, M. Taillades-jacquin, J. Rozie and D. J. Jones, Catalysis, 2018, 4660–4680 CAS
- W. Wannapakdee, T. Yutthalekha, P. Dugkhuntod, K. Rodponthukwaji, A. Thivasasith, S. Nokbin, T. Witoon, S. Pengpanich and C. Wattanakit, Catalysts, 2019, 9, 13 CrossRef
- L. E. Oi,
et al.
, RSC Adv., 2016, 6, 108741–108754 RSC
- M. Zhang, Z. Jin, Z. Zhang and H. Dang, Appl. Surf. Sci., 2005, 250, 29–34 CrossRef CAS
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9cc09715e |
| This journal is © The Royal Society of Chemistry 2020 |