
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTunable one-step double functionalization of graphene based on fluorographene chemistry†
Demetrios D.
Chronopoulos‡
 a,
Miroslav
Medveď‡
a,
Georgia
Potsi
a,
Ondřej
Tomanec
a,
Magdalena
Scheibe
a and
Michal
Otyepka
*ab
a,
Miroslav
Medveď‡
a,
Georgia
Potsi
a,
Ondřej
Tomanec
a,
Magdalena
Scheibe
a and
Michal
Otyepka
*ab
aFaculty of Science, Regional Centre of Advanced Technologies and Materials, Palacký University Olomouc, Šlechtitelů 27, CZ-771 46 Olomouc, Czech Republic
bDepartment of Physical Chemistry, Faculty of Science, Palacký University Olomouc, 17. listopadu 1192/12, 771 46 Olomouc, Czech Republic. E-mail: michal.otyepka@upol.cz
First published on 30th January 2020
Abstract
Double functionalized graphene derivatives were synthetized by a one-pot reaction of fluorographene with organometallic nucleophiles. Their nucleophilicity governed the preference for grafting and was utilized for tuning the functionalization. This approach paves the way toward the facile, up-scalable and controllable multifunctionalization of graphene.
Covalent modification of graphene1,2 significantly alters its electronic structure and physicochemical properties, changing the zero band-gap semimetal to an open gap semiconductor or even insulator,3,4 non-magnetic properties to diverse magnetic features, including ferro- or anti-ferromagnetism,5–7 and poor solubility to ready dispersibility in common polar solvents. However, the low reactivity of graphene and achieved low degree of functionalization remain obstacles against direct graphene functionalization. In the last decade, fluorographene8,9 (FG) has been launched as an alternative precursor for the preparation of covalently functionalized graphene derivatives bearing hydrocarbyl (alkyl, alkenyl, alkynyl and aryl),10–13 amino,14–16 cyano and carboxylic,17 hydroxyl,18,19 and sulfhydryl20–23 moieties. From a chemical point of view, FG behaves as an electrophile that reacts readily with nucleophiles,14,24 usually under mild conditions. It has been argued that the reactivity of FG is related to the occurrence of radical point defects,25 which trigger a cascade of nucleophilic substitution (SN) reaction along with simultaneous defluorination. Owing to the presence of highly electronegative fluorine atoms, the carbon lattice retains its electrophilic character, and thus its susceptibility to attack by nucleophiles, unless the content of fluorine is very low. As the substitution and defluorination proceed in a cascade manner, the resulting material usually has a well-defined stoichiometry and topology. The above properties make FG an excellent material for the high-yield, scalable, and controllable preparation of graphene derivatives.17,26,27 Moreover, the underlying sequential mechanism of SN reactions offers an opportunity to expand this class of materials by combining different nucleophilic agents in the reaction.
The multiple covalent functionalization of graphene is a very challenging task, paving the way toward multifunctional 2D materials. Hirsch et al. succeeded at the double functionalization of graphene via the consecutive treatment of graphenides with alkyl iodides and diazonium salts in a suspension.28 Besides bulk functionalization, they also studied single-side (monotopic) additions to CVD graphene and found that depending on the nature of the nucleophile, either double functionalization or retrofunctionalization could be realized. Our group reported a two-step double functionalization of graphene involving a photo-triggered Diels–Alder reaction on partially defluorinated FG.29 Both these previous works relied on two-step functionalization, i.e., two different moieties were grafted onto the graphene substrate in a consecutive way. However, if it were possible to perform multiple modifications in one step, the reaction time and wastage of material could be significantly reduced. Pennetreau et al. successfully performed the bi-functionalization of reduced graphene oxide sheets by using xanthates (radical precursors) and peroxides (initiators).30 Very recently, Lucherelli et al. reported the triple functionalization of graphene in a one-step reaction between aryl diazonium salts and graphenide.31 However, the abovementioned methods require harsh reaction conditions and highly reactive agents, which do not allow desirable control over the stoichiometry and topology of the target materials. Facile and controllable synthesis of multifunctionalized graphene thus remains a substantial challenge in the field of graphene chemistry.
In the present work, we exploited the susceptibility of FG toward nucleophiles as an alternative facile way for multiple modification of graphene avoiding the preparation of graphenides. Using this approach, we synthetized double covalently functionalized graphene derivatives in a one-pot reaction of FG with organometallic nucleophilic reagents. Two organolithium reagents containing alkyl (butyl – Bu) or heteroarene ring (thienyl – Th) moieties were reacted successfully with FG in one step to form a double functionalized graphene derivative. The sulfur of the Th moiety was used as a marker for obtaining information about the loading of the heteroarene unit onto graphene. In addition, control experiments and theoretical calculations were performed to provide insights into the concurrent double functionalization. Spectroscopy, thermogravimetry and microscopy techniques were applied to characterize the new graphene derivatives.
Organometallic reagents were chosen as nucleophiles since related Grignard reagents and/or lithium reagents have previously been successfully employed for the efficient covalent functionalization of FG and graphite fluoride (GF),10,11,32,33 providing highly functionalized graphene derivatives. Initially, FG layers were exfoliated by sonication of GF in anhydrous tetrahydrofuran (THF). Subsequently, different equivalents (eq.) of 2-thienyllithium (2-ThLi) were added dropwise to the suspension and the mixture was stirred under inert conditions for 18 h at room temperature to optimize conditions under which a sufficiently high number of fluorines remained on the prepared graphene derivative (see Section 1.2 and Table S1, ESI†). These fluorine atoms were necessary to ensure the presence of active sites on the partially modified FG for further functionalization. Note that the number of fluorines should be neither too large nor too small to achieve balanced functionalization by two different substituents. Otherwise, it would be difficult to obtain sufficient information about the process of double functionalization of FG. Based on X-ray photoelectron spectroscopy (XPS) data, we selected 2 eq. of 2-ThLi as the optimal amount for reaction with FG, retaining 12.4% of fluorine in the formed thienyl graphene (G-Th) (Fig. S1 and Table S1, ESI†). A similar amount of fluorine (14.9%) was detected when FG reacted with 2 eq. of butyl lithium (BuLi), forming butyl graphene (G-Bu) (Fig. S2 and Table S2, ESI†). According to XPS measurements, 4 eq. of organometallic reagent resulted in almost quantitative elimination of fluorine (Table S1, ESI†). In the next step, the possibility of one-step double functionalization of FG was pursued. In particular, reaction between FG and a mixture of 2 eq. of 2-ThLi and 2 eq. of BuLi was carried out (Fig. 1). The prepared butyl thienyl graphene (G-Th/Bu) contained 2.6% and 2.7% of fluorine and sulfur, respectively (Table S2, ESI†). Thus, the sulfur content was significantly decreased compared to the detected amount of sulfur (9.7%) in G-Th, for which the same quantity of 2-ThLi was used (Table S2, ESI†), while defluorination was practically complete. These findings indicate that (i) there was a strong preference for the grafting of Bu groups, and (ii) there was a synergic reductive effect of both nucleophiles, resulting in a higher degree of defluorination compared to that in independent full “BuLi + FG” and partial “2-ThLi + FG” reactions. These observations may be due to the higher reactivity of the Bu nucleophile in a competitive substitution reaction or from a retroreaction of BuLi with G-Th, in which some of the Th moieties are removed and replaced by Bu groups.
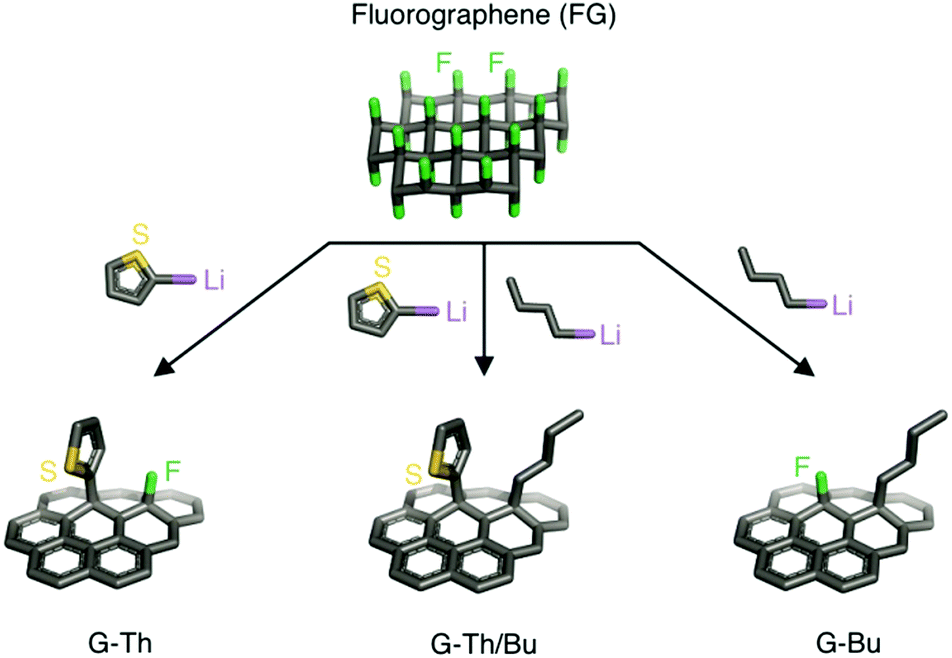 | ||
| Fig. 1 Overview of single (left and right) and double (middle) functionalization on FG. | ||
To investigate the above scenarios, two experiments were performed. In the first, FG was reacted with 2 eq. of 2-ThLi, and then after 18 h of stirring under inert conditions, 2 eq. of BuLi were added. The XPS spectrum of the product (Fig. S3, ESI†) corresponded to thienyl butyl graphene (G-Th(Bu)) containing 10.1% sulfur, i.e., a value close to that of G-Th. A much lower content of fluorine (2.6%) accompanied by an increased proportion of carbon indicated attachment of Bu groups onto pre-formed G-Th, substituting the remaining fluorine atoms, without any retrofunctionalization of the Th groups. As an opposite control, 2 eq. of BuLi were mixed with a suspension of FG and then after 18 h of stirring, 2 eq. of 2-ThLi were added, forming G-Bu(Th). In this case, the detected amount of sulfur was found to be 0.8%, showing that only a few Th groups were attached to the pre-formed G-Bu. The amount of fluorine was similar to that in G-Bu, indicating that no further nucleophilic substitution or reductive defluorination took place, i.e., the reaction was practically stopped. Note that high-resolution S2p XPS spectra of all samples containing Th groups consistently exhibited two characteristic peaks at ∼164.0 eV and ∼165.3 eV, corresponding to S2p3/2 and S2p1/2, respectively, of a sulfur atom embedded in the Th ring (Fig. S4–S7, ESI†).
Double functionalization of FG was also confirmed by Fourier transform infrared (FT-IR) spectroscopy (Fig. 2). Two characteristic bands at around 1580 and 1460 cm−1 appeared in FT-IR spectra of the graphene derivatives, indicating the formation of conjugated C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C double bonds due to the elimination of fluorines of FG34 by nucleophilic substitution and concurrent reduction. The FT-IR spectrum of G-Th/Bu displayed a band at 2960–2850 cm−1, attributed to aliphatic C–H stretching vibrations of the Bu groups, and three peaks at 692, 790 and 1074 cm−1, associated with the presence of thiophene rings.35 The same peaks appeared in all G-Th derivatives but with different intensities, reflecting the amount of grafted thiophene (Fig. S8, ESI†). Hence, the spectrum of G-Bu(Th) was similar to that of G-Bu because of a very low amount of Th groups grafted onto graphene, in line with the XPS data.
C double bonds due to the elimination of fluorines of FG34 by nucleophilic substitution and concurrent reduction. The FT-IR spectrum of G-Th/Bu displayed a band at 2960–2850 cm−1, attributed to aliphatic C–H stretching vibrations of the Bu groups, and three peaks at 692, 790 and 1074 cm−1, associated with the presence of thiophene rings.35 The same peaks appeared in all G-Th derivatives but with different intensities, reflecting the amount of grafted thiophene (Fig. S8, ESI†). Hence, the spectrum of G-Bu(Th) was similar to that of G-Bu because of a very low amount of Th groups grafted onto graphene, in line with the XPS data.
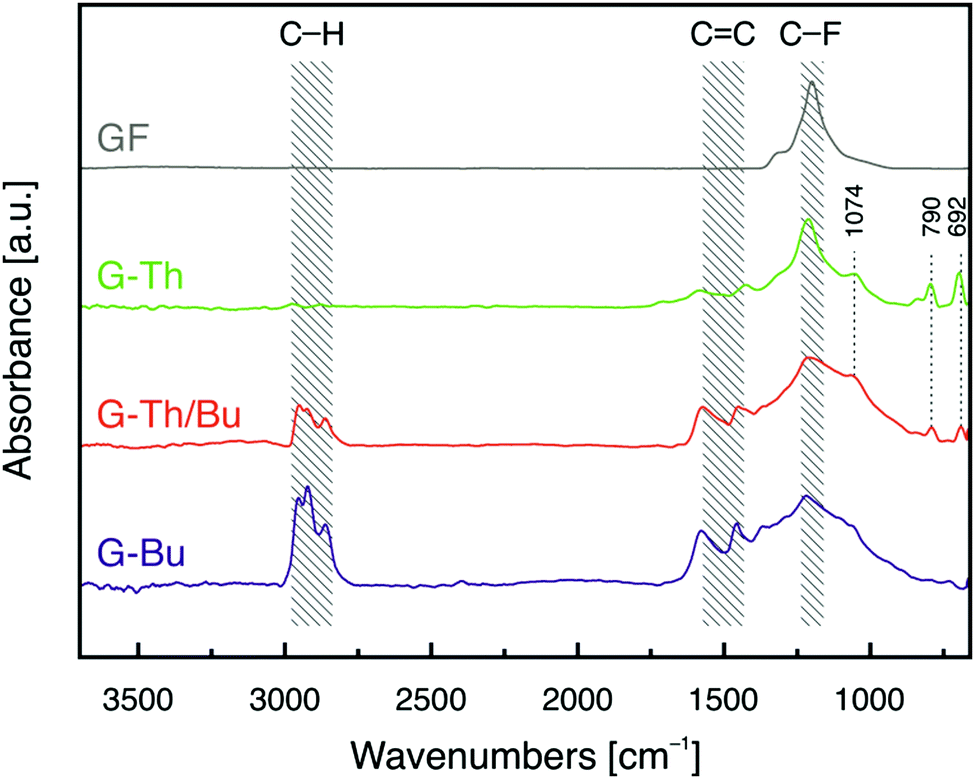 | ||
| Fig. 2 FT-IR spectra of single and double functionalized graphene derivatives (G-Th, G-Bu and G-Th/Bu) and pristine GF. | ||
Raman spectroscopy was employed to further confirm the formation of graphene derivatives via one-step as well as successive double functionalization of FG. All products exhibited characteristic D and G bands at around 1360 and 1590 cm−1, respectively, in contrast to Raman inactive pristine GF (Fig. S9–S14, ESI†). The G-band was assigned to vibrations of sp2 carbon domains formed by the reductive defluorination of FG, whereas the D-band was attributed to vibrations involving sp3 hybridized carbons. Moreover, the Raman spectra of Th-containing derivatives G-Th/Bu and G-Th(Bu) displayed a characteristic peak due to Th at around 1450 cm−1,36 confirming successful grafting of this moiety (Fig. S12 and S13, ESI†). In the case of G-Bu(Th), this band was less evident due to the low amount of Th in G-Bu(Th) (Fig. S14, ESI†). Information about the thermal stability of the prepared derivatives was obtained by thermogravimetric analysis (TGA) under N2 (Fig. S15 and S17, ESI†). The precursor GF is stable up to 400 °C and loses ca. 75% of its weight over the temperature range 450–650 °C due to cleavage of C–F bonds and CXFY fragments.37 The mass of G-Th/Bu slowly decreased to ca. 65% between 180 and 530 °C due to detachment and/or decomposition of the Bu and Th moieties. The detachment of these units was verified by mass spectroscopy analysis of evolving gases. Mass fragments with m/z of 39 and 58 were attributed to Th, whereas that with m/z of 43 to Bu (Fig. S16, ESI†), demonstrating successful covalent binding of both units onto the graphene lattice. The similar shape of the decomposition curves of G-Bu(Th) and G-Bu also corroborated that both derivatives mainly contained Bu units (Fig. S17, ESI†). Transmission electron microscopy (TEM) (Fig. 3a and Fig. S18–S20, ESI†) and atomic force microscopy (AFM) (Fig. 3b and c) verified that all the fabricated materials were composed of few-layer flakes. Energy dispersive spectroscopy (EDS) elemental mapping of G-Th/Bu proved the presence of S and revealed its uniform distribution on the surface (Fig. 3d–f).
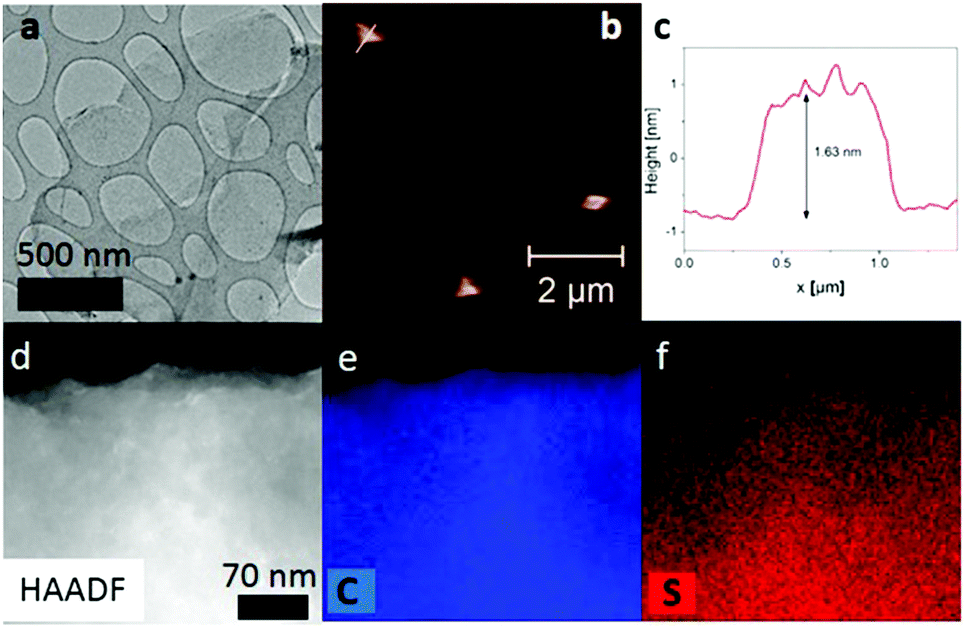 | ||
| Fig. 3 (a) HR-TEM, (b and c) AFM image and height profile, and (d–f) dark field image with carbon and sulfur EDS maps of G-Th/Bu flakes. | ||
To rationalize the experimental observations, we performed density functional theory (DFT) calculations of nucleophilic strengths and binding energies of nucleophiles on different types of partially functionalized FG (pFG) substrates by applying the ωB97X-D method38 with the 6-31+G(d) basis set.39 Solvent effects were included by using the universal continuum solvation model based on solute electron density (SMD).40 All calculations were performed with the Gaussian09 program.41 Comparison of the natural charges on terminal carbon atoms revealed that the Bu anion was a notably stronger nucleophilic agent than the Th anion (Fig. 4a and b). This is one of the key factors determining the binding capabilities of these two agents on FG substrates. Whereas attachment of a Bu anion on pFG, butylated pFG (G-Bu) and pFG functionalized by thienyl groups (G-Th) was energetically favorable, binding of the Th anion on these substrates was much less favored (Fig. 4c–e). This is in line with the experimental findings for the sequential functionalization, which was found to be feasible only if the weaker nucleophile (i.e., the Th anion) was used in the first step (the G-Th(Bu) case in Table S2, ESI†). In this case, the first phase of the reaction led to formation of a G-Th substrate, which was then susceptible to further attack by the Bu anion with a binding energy of −1.7 kcal mol−1 in THF (Fig. 4e). In the reverse case (G-Bu(Th)), a Th anion could hardly attach to the G-Bu substrate formed in the first phase of the reaction (ΔE = 21.6 kcal mol−1 in THF, Fig. 4d). Therefore, the reaction essentially stopped at this stage. However, it could proceed if a stronger nucleophile were added (G-Bu). Evidently, the binding energies follow the electrophilic strength of the substrate, decreasing in the order pFG > G-Bu > G-Th (cf. partial charges on defluorinated carbon atoms in Fig. S21, ESI†), in keeping with the electron withdrawing/donating strengths of the substituents. Finally, sterical hindrance among addends at the final phase of the reaction may put limitations on the degree of functionalization (Table S3, ESI†).
 | ||
| Fig. 4 Natural charges on terminal carbon atoms of (a) n-butyl carbanion and (b) thiophenyl anion, and optimized model structures of the graphene substrate partially functionalized by (c) solely fluorine adatoms, (d) n-butyl (Bu) and (e) thiophenyl (Th) groups in the vicinity of a defluorinated region (the carbon atom attacked by a nucleophile in the reaction is encircled). Binding energies (in kcal mol−1) of the nucleophiles attacking the respective substrates in the gas phase (blue) and THF (red) were obtained at the ωB97X-D/6-31+G(d)/SMD level of theory. | ||
In summary, an efficient and straightforward approach to prepare double functionalized graphene derivatives taking advantage of the susceptibility of FG toward reduction and (multiple) nucleophilic substitutions was described. The possibility of using either a sequential or one-step reaction enables the controllable and scalable preparation of multiple modified graphene derivatives. Theoretical calculations and experiments showed that grafting of the units onto FG is driven by their nucleophilicity and the electrophilicity of the substrate, thus providing helpful guidelines for the controllable double/multiple functionalization.
We acknowledge support from MEYS (CZ.02.1.01/0.0/0.0/16_019/0000754) and the ERC (683024 from the H2020). We thank M. Petr for XPS, J. Ugolotti for TGA, C. Perez-Reyes for TEM, and K. Štymplová for Raman spectra measurements.
Conflicts of interest
There are no conflicts to declare.References
- G. Bottari, M. Á. Herranz, L. Wibmer, M. Volland, L. Rodríguez-Pérez, D. M. Guldi, A. Hirsch, N. Martín, F. D’Souza and T. Torres, Chem. Soc. Rev., 2017, 46, 4464–4500 RSC.
- V. Georgakilas, M. Otyepka, A. B. Bourlinos, V. Chandra, N. Kim, K. C. Kemp, P. Hobza, R. Zboril and K. S. Kim, Chem. Rev., 2012, 112, 6156–6214 CrossRef CAS PubMed.
- G. Lu, K. Yu, Z. Wen and J. Chen, Nanoscale, 2013, 5, 1353–1368 RSC.
- N. T. T. Tran, D. K. Nguyen, O. E. Glukhova and M.-F. Lin, Sci. Rep., 2017, 7, 1–13 CrossRef PubMed.
- J. Zhou, Q. Wang, Q. Sun, X. S. Chen, Y. Kawazoe and P. Jena, Nano Lett., 2009, 9, 3867–3870 CrossRef CAS PubMed.
- J. Tuček, P. Błoński, Z. Sofer, P. Šimek, M. Petr, M. Pumera, M. Otyepka and R. Zbořil, Adv. Mater., 2016, 28, 5045–5053 CrossRef PubMed.
- R. Langer, P. Błoński and M. Otyepka, Phys. Chem. Chem. Phys., 2019, 21, 12697–12703 RSC.
- D. D. Chronopoulos, A. Bakandritsos, M. Pykal, R. Zbořil and M. Otyepka, Appl. Mater. Today, 2017, 9, 60–70 CrossRef PubMed.
- W. Feng, P. Long, Y. Feng and Y. Li, Adv. Sci., 2016, 3, 1500413 CrossRef PubMed.
- D. D. Chronopoulos, A. Bakandritsos, P. Lazar, M. Pykal, K. Čépe, R. Zbořil and M. Otyepka, Chem. Mater., 2017, 29, 926–930 CrossRef CAS PubMed.
- V. Mazánek, A. Libánská, J. Šturala, D. Bouša, D. Sedmidubský, M. Pumera, Z. Janoušek, J. Plutnar and Z. Sofer, Chem. – Eur. J., 2017, 23, 1956–1964 CrossRef PubMed.
- D. D. Chronopoulos, M. Medved’, P. Błoński, Z. Nováček, P. Jakubec, O. Tomanec, A. Bakandritsos, V. Novotná, R. Zbořil and M. Otyepka, Chem. Commun., 2019, 55, 1088–1091 RSC.
- W. Lai, J. Liu, L. Luo, X. Wang, T. He, K. Fan and X. Liu, Chem. Commun., 2018, 54, 10168–10171 RSC.
- K. E. Whitener, R. Stine, J. T. Robinson and P. E. Sheehan, J. Phys. Chem. C, 2015, 119, 10507–10512 CrossRef CAS.
- C. Bosch-Navarro, M. Walker, N. R. Wilson and J. P. Rourke, J. Mater. Chem. C, 2015, 3, 7627–7631 RSC.
- X. Ye, L. Ma, Z. Yang, J. Wang, H. Wang and S. Yang, ACS Appl. Mater. Interfaces, 2016, 8, 7483–7488 CrossRef CAS PubMed.
- A. Bakandritsos, M. Pykal, P. Błoński, P. Jakubec, D. D. Chronopoulos, K. Poláková, V. Georgakilas, K. Čépe, O. Tomanec, V. Ranc, A. B. Bourlinos, R. Zbořil and M. Otyepka, ACS Nano, 2017, 11, 2982–2991 CrossRef CAS PubMed.
- P. Gong, J. Wang, W. Sun, D. Wu, Z. Wang, Z. Fan, H. Wang, X. Han and S. Yang, Nanoscale, 2014, 6, 3316–3324 RSC.
- J. Tuček, K. Holá, A. B. Bourlinos, P. Błoński, A. Bakandritsos, J. Ugolotti, M. Dubecký, F. Karlický, V. Ranc, K. Čépe, M. Otyepka and R. Zbořil, Nat. Commun., 2017, 8, 14525 CrossRef PubMed.
- V. Urbanová, K. Holá, A. B. Bourlinos, K. Čépe, A. Ambrosi, A. H. Loo, M. Pumera, F. Karlický, M. Otyepka and R. Zbořil, Adv. Mater., 2015, 27, 2305–2310 CrossRef PubMed.
- P. Kovaříček, Z. Bastl, V. Valeš and M. Kalbac, Chem. – Eur. J., 2016, 22, 5404–5408 CrossRef PubMed.
- A. Stathis, I. Papadakis, N. Karampitsos, S. Couris, G. Potsi, A. B. Bourlinos, M. Otyepka and R. Zboril, ChemPlusChem, 2019, 84, 1288–1298 CrossRef CAS PubMed.
- J. Sturala, S. Hermanová, L. Artigues, Z. Sofer and M. Pumera, Nanoscale, 2019, 11, 10695–10701 RSC.
- M. Dubecký, E. Otyepková, P. Lazar, F. Karlický, M. Petr, K. Čépe, P. Banáš, R. Zbořil and M. Otyepka, J. Phys. Chem. Lett., 2015, 6, 1430–1434 CrossRef PubMed.
- M. Medveď, G. Zoppellaro, J. Ugolotti, D. Matochová, P. Lazar, T. Pospíšil, A. Bakandritsos, J. Tuček, R. Zbořil and M. Otyepka, Nanoscale, 2018, 10, 4696–4707 RSC.
- A. Bakandritsos, D. D. Chronopoulos, P. Jakubec, M. Pykal, K. Čépe, T. Steriotis, S. Kalytchuk, M. Petr, R. Zbořil and M. Otyepka, Adv. Funct. Mater., 2018, 28, 1801111 CrossRef.
- D. Matochová, M. Medved’, A. Bakandritsos, T. Steklý, R. Zbořil and M. Otyepka, J. Phys. Chem. Lett., 2018, 9, 3580–3585 CrossRef PubMed.
- K. C. Knirsch, R. A. Schäfer, F. Hauke and A. Hirsch, Angew. Chem., Int. Ed., 2016, 55, 5861–5864 CrossRef CAS PubMed.
- H. Barès, A. Bakandritsos, M. Medveď, J. Ugolotti, P. Jakubec, O. Tomanec, S. Kalytchuk, R. Zbořil and M. Otyepka, Carbon, 2019, 145, 251–258 CrossRef.
- F. Pennetreau, O. Riant and S. Hermans, Chem. – Eur. J., 2014, 20, 15009–15012 CrossRef CAS PubMed.
- M. A. Lucherelli, J. Raya, K. F. Edelthalhammer, F. Hauke, A. Hirsch, G. Abellán and A. Bianco, Chem. – Eur. J., 2019, 25, 13218–13223 CrossRef CAS PubMed.
- I. Papadakis, D. Kyrginas, A. Stathis, S. Couris, G. Potsi, A. B. Bourlinos, O. Tomanec, M. Otyepka and R. Zboril, J. Phys. Chem. C, 2019, 123, 25856–25862 CrossRef CAS.
- K. A. Worsley, P. Ramesh, S. K. Mandal, S. Niyogi, M. E. Itkis and R. C. Haddon, Chem. Phys. Lett., 2007, 445, 51–56 CrossRef CAS.
- R. Zbořil, F. Karlický, A. B. Bourlinos, T. A. Steriotis, A. K. Stubos, V. Georgakilas, K. Šafářová, D. Jančík, C. Trapalis and M. Otyepka, Small, 2010, 6, 2885–2891 CrossRef PubMed.
- A. Stergiou, D. K. Perivoliotis and N. Tagmatarchis, Nanoscale, 2019, 11, 7335–7346 RSC.
- M. Melucci, E. Treossi, L. Ortolani, G. Giambastiani, V. Morandi, P. Klar, C. Casiraghi, P. Samorì and V. Palermo, J. Mater. Chem., 2010, 20, 9052–9060 RSC.
- J. T. Robinson, J. S. Burgess, C. E. Junkermeier, S. C. Badescu, T. L. Reinecke, F. K. Perkins, M. K. Zalalutdniov, J. W. Baldwin, J. C. Culbertson, P. E. Sheehan and E. S. Snow, Nano Lett., 2010, 10, 3001–3005 CrossRef CAS PubMed.
- J.-D. Chai and M. Head-Gordon, Phys. Chem. Chem. Phys., 2008, 10, 6615–6620 RSC.
- R. Ditchfield, W. J. Hehre and J. A. Pople, J. Chem. Phys., 1971, 54, 724–728 CrossRef CAS.
- A. V. Marenich, C. J. Cramer and D. G. Truhlar, J. Phys. Chem. B, 2009, 113, 6378–6396 CrossRef CAS PubMed.
- M. J. Frisch, et al., Gaussian09.D01, Gaussian Inc., Wallingford, CT, 2009 Search PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9cc09514d |
| ‡ Both authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2020 |