
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAccess to the most sterically crowded anilines via non-catalysed C–C coupling reactions†
Jan
Vrána
 *,
Maksim A.
Samsonov
,
Vlastimil
Němec
and
Aleš
Růžička
*,
Maksim A.
Samsonov
,
Vlastimil
Němec
and
Aleš
Růžička
Department of General and Inorganic Chemistry, Faculty of Chemical Technology, University of Pardubice, Studentská 573, CZ-532 10, Pardubice, Czech Republic. E-mail: jan.vrana@upce.cz
First published on 21st January 2020
Abstract
Variously substituted 2,6-bis(1,1-diarylethyl)anilines and 2,6-bis(trityl)anilines were prepared by a three-step high-yield process. Dimethyl-2-aminoisophtalate was modified by reaction with arylmagnesium bromides, and the hydroxy-derivatives obtained were etherified. Under the non-catalysed C–C coupling protocol, the formed bis[methyl(methoxy)diaryl]anilines react with various Grignard reagents to give highly substituted products. The buried volumes around the central nitrogen atom of the prepared compounds exceed the parameters for the known most sterically hindered anilines by about 20%.
The kinetic stabilisation of the reactive metal centres in terms of ligand exchange or oxidation plays an important role in many branches of synthetic chemistry. Sterically demanding ligands, which provide such a kind of stabilisation, have accessed new classes of compounds for example in main-group chemistry by introducing compounds in an unusual oxidation state, which can adopt some properties of transition metals and are therefore active in homogeneous catalysis or small-molecule activation.1 Next, the so-called double-bond rule, which does not allow the formation of multiple bonds between the elements of the third period or higher, has been broken by the synthesis of various heavier alkene analogues.2 Moreover, bulky ligands are essential in homogeneous catalysis, e.g. the second and third generation of Grubbs catalysts.3 A considerable part of these modern ligands is based on aniline moieties, which tune their steric properties. Anilines are the core of various sterically demanding ligands, e.g. N-heterocyclic carbenes, β-diketiminates, amidinates, guanidinates and others.4 They have also been used many times as ligands themselves in the form of amides or imides.3,5
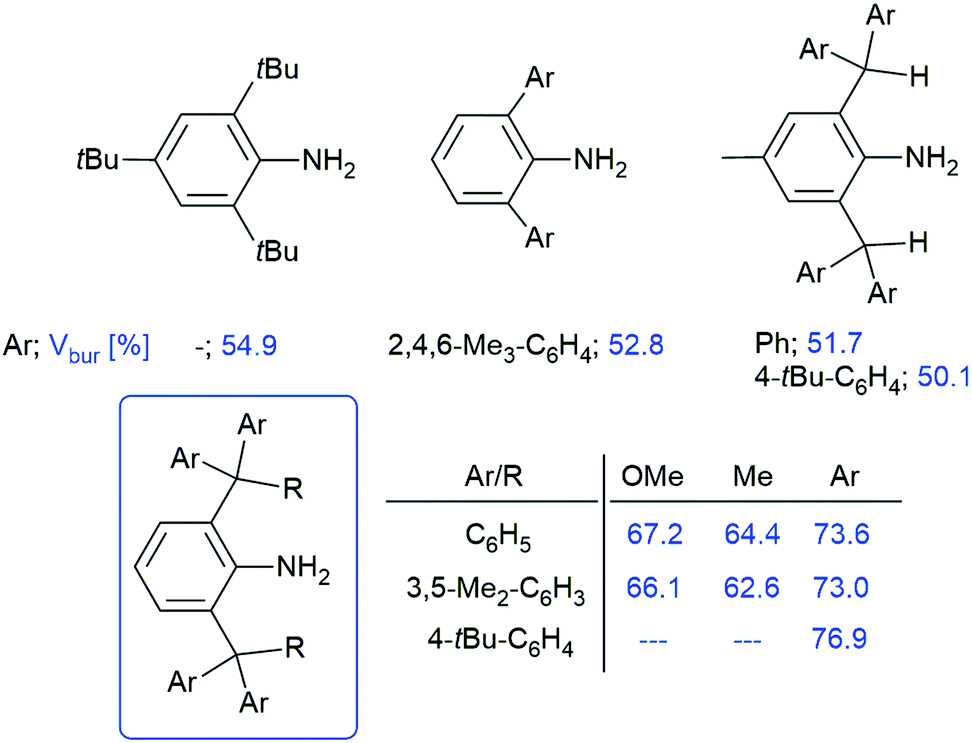
A large variety of anilines with different backbones have been prepared in the past few decades. The steric properties of anilines, unlike those of aliphatic amines, are very easily tuneable by changing the substituents in the neighbouring positions. Anilines containing a terphenyl backbone (Fig. 1A) are accessible through the reduction of the corresponding azide and have great shielding provided by the ortho-aryl substituents, which enables the stabilisation of extremely reactive species such as biradicaloids.6,7 In the past few decades, 2,6-bis(benzhydryl)anilines (Fig. 1B) have been the most extensively studied group of sterically demanding anilines. They can be smoothly prepared by melting diphenylmethanol and the appropriate aniline in the presence of concentrated HCl and zinc chloride on a large scale (>60 g). The steric bulk of 2,6-bis(benzhydryl)anilines can be further increased by the use of substituted benzhydrols, e.g. (3,5-tBu2-C6H3)2C(H)OH or (4-tBu-C6H4)2C(H)OH.8 The kinetic stabilisation provided by these anilines was demonstrated by Bertrand, who published the first compound containing a terminal phosphorus atom bonded to a main group element centre in the form of a phosphino-phosphinidene.8 One of the limitations of 2,6-(benzhydryl)anilines is the potential activation of a rather acidic C–H bond in the CHAr2 moiety.9 Nevertheless, there is no published procedure that would lead to the virtual replacement of this hydrogen atom by any other organic substituent. Simple electrophilic alkylation of anilines by Ph2RC(OH), which was mentioned above, results in N-alkylation or exhibits no reaction at all.10 This fact makes 2,4,6-tri-tert-butylaniline (Fig. 1C), one of the oldest examples of a sterically demanding aniline, the only aniline with both ortho-positions occupied by a tertiary organic group. Interestingly, it is the only amine that can stabilize iminochlorophosphane Ar–N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) P–Cl in its monomeric form even in the solid state, while slightly smaller substituents such as 2,6-bis-(2,4,6-trimethylphenyl)phenyl or 2,6-diisopropylphenyl allow dimerisation to 1,3-dichloro-cyclo-diphosphadiazanes, [ClP(μ-NAr)]2.11,12 No aniline bearing a different tertiary organic group other than tBu has been published even though there has been a high demand for bulky ligands in the last decade. Therefore, we present a simple, high-yielding synthesis of a series of sterically demanding anilines bearing CAr3 and CAr2Me substituents in the ortho- positions.
P–Cl in its monomeric form even in the solid state, while slightly smaller substituents such as 2,6-bis-(2,4,6-trimethylphenyl)phenyl or 2,6-diisopropylphenyl allow dimerisation to 1,3-dichloro-cyclo-diphosphadiazanes, [ClP(μ-NAr)]2.11,12 No aniline bearing a different tertiary organic group other than tBu has been published even though there has been a high demand for bulky ligands in the last decade. Therefore, we present a simple, high-yielding synthesis of a series of sterically demanding anilines bearing CAr3 and CAr2Me substituents in the ortho- positions.
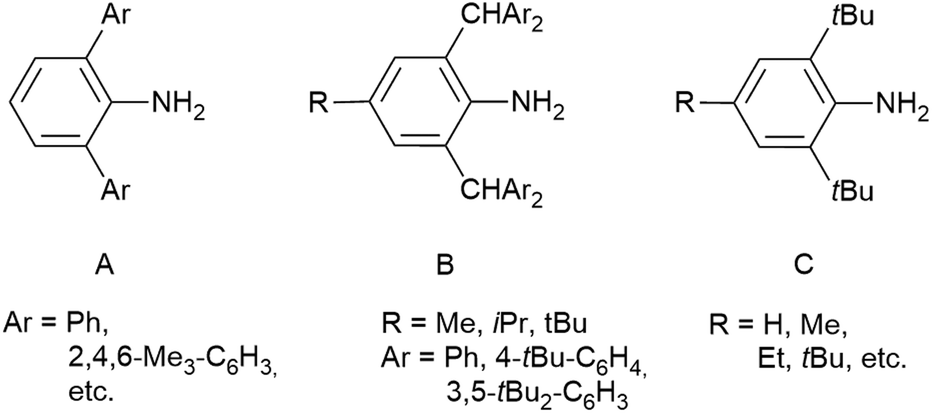 | ||
| Fig. 1 Examples of known bulky anilines. | ||
The synthesis of anilines derived from 2,6-benzhydrylanilines (Fig. 1B) by the virtual replacement of the hydrogen atoms by different functional groups could not be achieved by direct alkylation of anilines. Therefore, we have modified the synthetic procedure for the preparation of anilines 1-NH2-2,6-(CH(CH2R)2)2-C6H3, where R = Me, Et, nPr, which were used as building blocks for sterically hindered N-heterocyclic carbenes.13 Dimethyl-2-aminoisophtalate reacts with an overstoichiometric amount of the appropriate aryl-Grignard reagent (Scheme 1), giving anilines 1-NH2-2,6-[C(OH)Ar2]2-C6H3 (1a–c) after hydrolysis. When compared to the direct alkylation mentioned above, this method leads directly to anilines bearing functionalised benzhydryl groups.
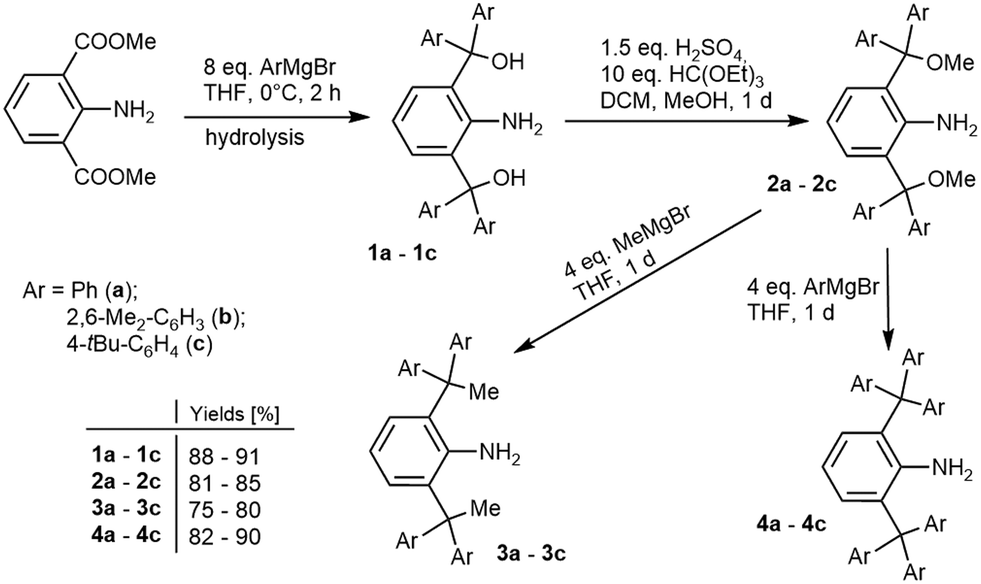 | ||
| Scheme 1 Synthesis of bulky anilines. | ||
Moreover, these reactions can be performed on a large scale with high yields (∼90%). However, the hydroxy-groups are not suitable for subsequent C–C coupling. Therefore, they have been converted to a methoxy group by etherification with methanol in the presence of sulphuric acid and triethyl orthoformate. Triethyl orthoformate is not essential for the reaction, but it shortens the reaction time from two weeks to one day. The treatment of 2a–c with methyl/arylmagnesium bromide (Scheme 1) led to the replacement of the methoxy groups with a methyl/aryl group, giving anilines 3a–c/4a–c. Analogical Kumada–Corriu-type coupling requires catalysis by various Ni-or Pd-catalysts.14 Our coupling reactions proceed rapidly (full conversion after one day) giving high yields (75–90% of isolated yield) under mild conditions without a catalyst. Moreover, this is the first coupling reaction of the bulky triaryl-moiety substituted with a rather unreactive methoxy-group. A similar alkylation procedure was performed only in the cases of 2-methoxymethyl-6-methylaniline and 2-methoxymethylaniline, which were alkylated by vinyl- and allylmagnesium bromides.15
The reaction mechanism of these reactions was investigated by Mann and Stewart in 1954 and later confirmed by Görl and Alt in 2007.16,17
In our system, the mechanism is suggested to be analogical, despite the system being more complex. In the first step (Scheme 2, step A), the amine group is deprotonated by the first equivalent of the Grignard reagent. The coordination of the magnesium atom with the methoxy group weakens the C–O bond (Scheme 2, step B), which is cleaved and the methoxymagnesium bromide is eliminated. The resulting dearomatized imine is less stable when compared to the 1,4-dipolar tautomeric form, which reacts with the second equivalent of the Grignard reagent (Scheme 2, step C). The steps B and C repeat again replacing the second methoxy-group with a methyl or aryl group. The hydrolysis of the final product leads to the desired anilines 3a–c and 4a–c. All prepared compounds were isolated as colourless solids soluble in THF, aromatic and chlorinated solvents with the exception of 4a, which is only sparingly soluble in chlorinated solvents. The 1H and 13C NMR spectra of the prepared anilines exhibited one set of expected signals. The signals of amine groups in the 1H spectra of 1a–c (∼4.0 ppm) and 2a–c (∼4.9 ppm) were shifted downfield in comparison with 3a–c and 4a–c (∼3.5 ppm), which indicates the presence of the hydrogen bond NH⋯O. Similarly, the signals of the hydroxy groups in 1a–c were also shifted downfield with respect to analogous Ph3COH (2.78 ppm), indicating the presence of OH⋯N hydrogen bonding.18
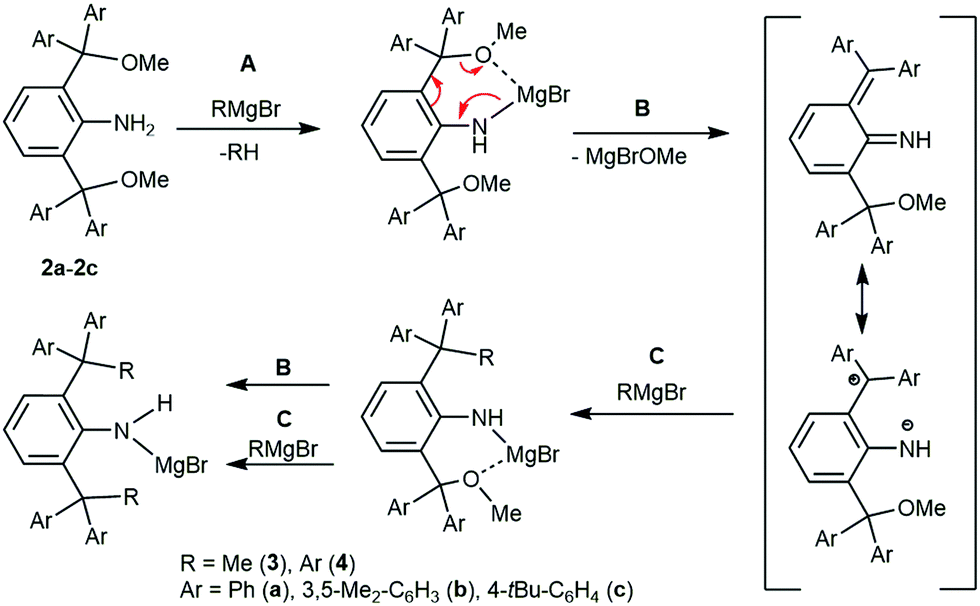 | ||
| Scheme 2 Synthesis of bulky anilines, C–C coupling. | ||
Despite the diversity of the substituents on the benzhydryl moieties, the molecular structures of 1a–4a, 1b–4b and 4c are analogous (see Fig. 2 and 3 for examples; for further information, see the ESI†). Expectedly, the intramolecular H-bonding is promoted within the series of 1 and 2 (see the ESI†). In the series of 3 and 4, significant steric shielding by hydrocarbon moieties did not allow stronger intermolecular contacts within the crystal lattice. In structures of the methoxy or trityl-substituted compounds of series 2 and 4, the C–N bond is much shorter (1.370(3)–1.373(4) and 1.377(5)–1.392(3) Å) than that in the structures with hydroxyl groups (series 1 (1.407(4)–1.422(2) Å)) or diarylethyl-substituted compounds of series 3 (1.405(1)–1.412(5) Å). Surprisingly enough, the only compound with the anti-orientation of the oxygen atoms is 1a (Fig. 2).
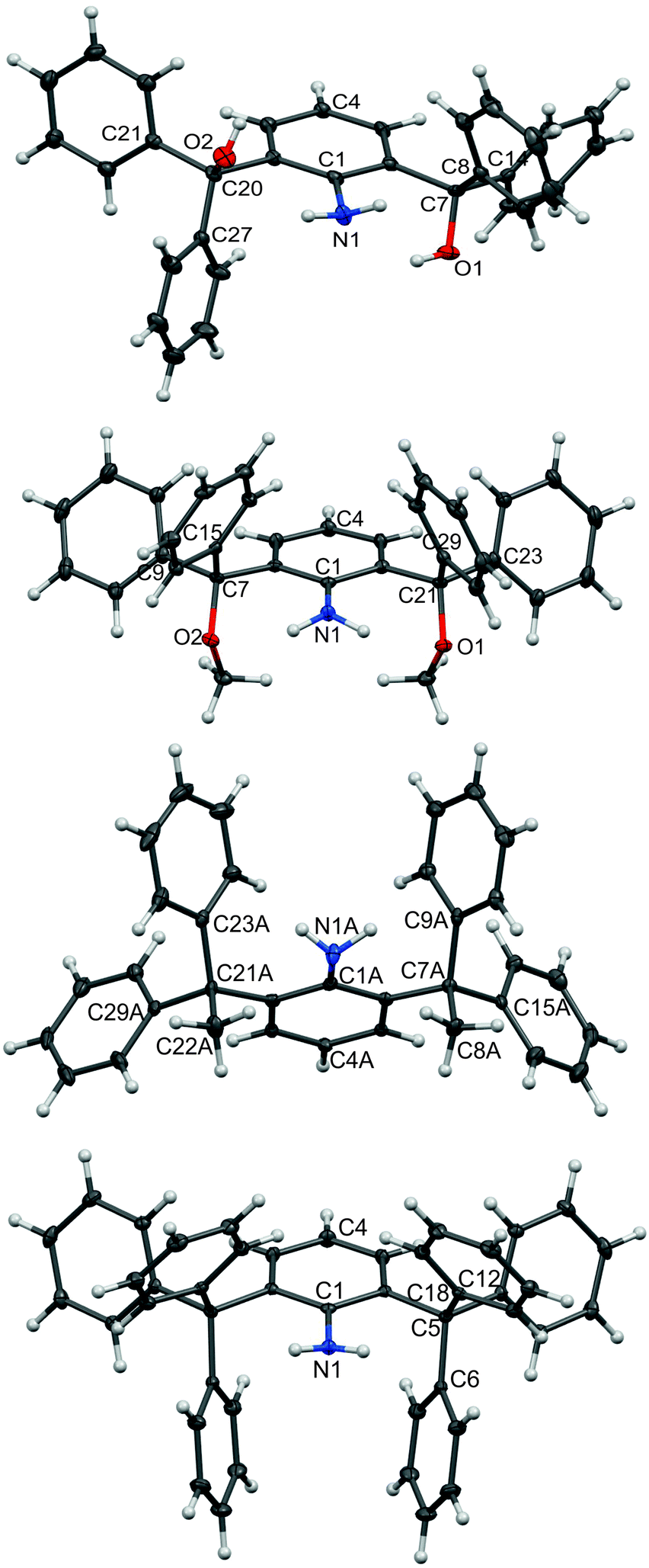 | ||
| Fig. 2 The molecular structures of 1a, 2a, 3a and 4a. ORTEP diagrams, 35–50% probability level; solvent molecules have been omitted for clarity. Selected interatomic distances [Å]: 1a: O1 N1 2.830(5), O2 N1 2.891(5), C7 C20 5.106(5), C1 N1 1.407(4); 2a: O1 N1 2.907(4), O2 N1 2.866(4), C7 C21 5.097(4), C1 N1 1.373(4); 3a: (the second independent molecule is omitted for clarity, its parameters are given in parentheses): C8A N1A 3.082(6) (3.087(6)), C22A N1A 3.018(6) (2.995(6)), C7A C21A 5.159(5) (5.176(5)), C1A N1A 1.412(5) (1.411(5)); 4a: C1 N1 1.392(3), C5 C5i 5.148(4). | ||
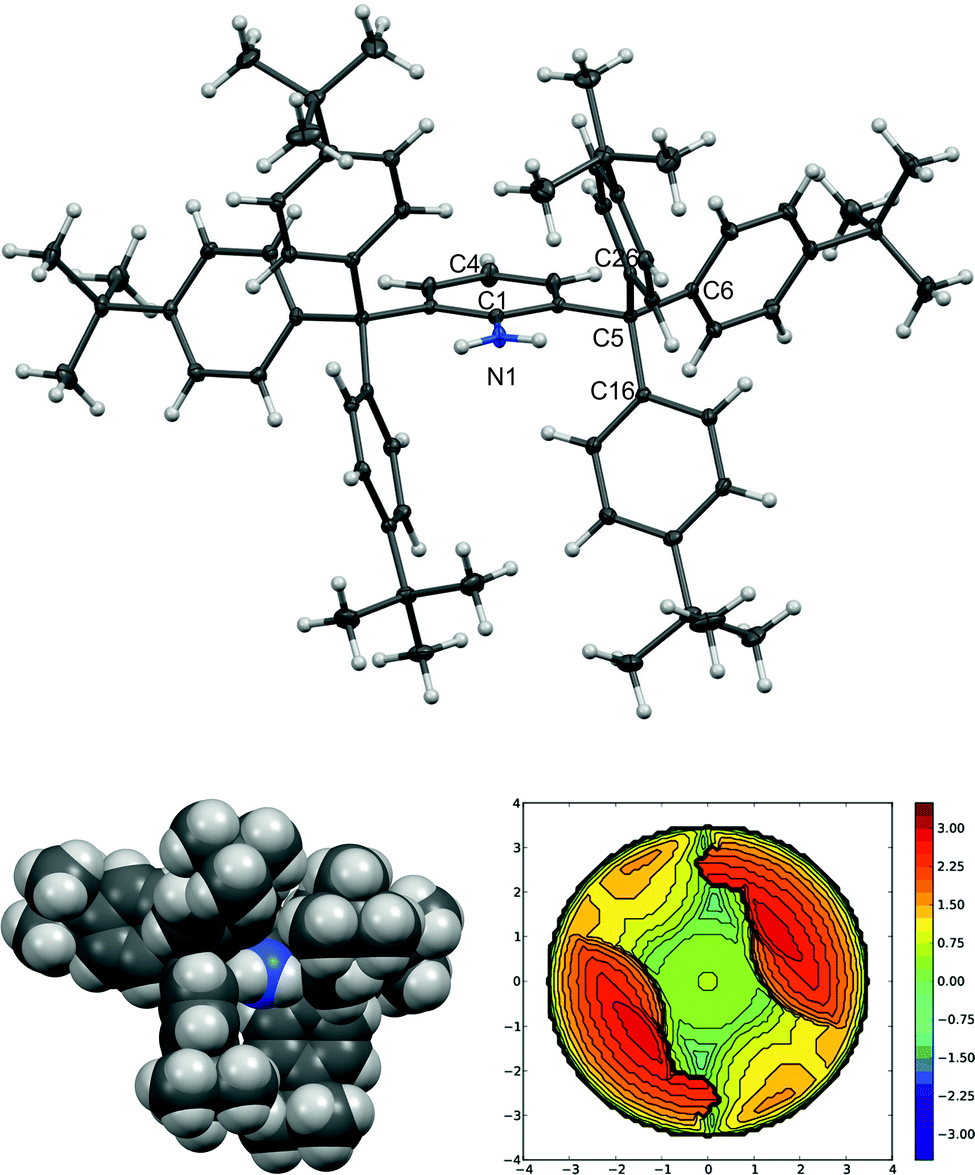 | ||
| Fig. 3 The molecular structure of 4c, ORTEP diagram (left), 40% probability level, space-filling model (middle), the dichloromethane solvate molecule has been omitted for clarity. Selected interatomic distances [Å]: C5 N1 2.929(3), C5 C5i 5.193(5), C1 N1 1.377(5). A steric map of 4c (right). | ||
Within series 4, the sterical hindrance changes resonate with the differences in the interplanar angles between the plane of the middle (aniline) ring and the planes defined by three pivotal carbon atoms (for example C6 C12 and C18 for 4a) of the phenyl rings. These values range from the ideal perpendicular value found for 4a, to 84.44° and 88.41° in the case of 4b, to a rather unsymmetrical situation in the case of 4c (80.66° and 89.67°).
In order to quantify the steric shielding of the ortho-substituents, we calculated buried volumes, i.e. the percentage of the volume of a sphere centred around the nitrogen atom with a radius of 3.5 Å, which is occupied by the bulky aryl backbone (Scheme 3).19 For comparison, we also calculated the buried volumes of the other published sterically demanding anilines (Scheme 3) mentioned above (Fig. 1). The anilines 4a–c exhibit the highest percentage of the buried volume, which is provided by the bulky trityl moieties. This percentage decreases in the order 4a–c > 2a/2b > 3a/3b > 1a/1b, but all prepared anilines exhibit similar or higher values than the bulkiest crystallographically characterised anilines published to date.20 This fact has also been visualised by steric maps (Fig. 3; for more information, see ESI†), which document the effectivity of the steric shielding around the central nitrogen atom. Interestingly, the anilines 1a–4a exhibit a slightly higher percentage of buried volume than the series 1b–4b, which indicates the less influence of the derivatisation of the phenyl substituents of the benzhydryl groups on the steric hindrance around the nitrogen atom. A similar trend could also be observed in the case of published 2,6-benzhydryl-anilines (Scheme 3).
 | ||
| Scheme 3 Buried volumes of published (upper part) and prepared anilines. | ||
In summary, the most sterically demanding 2,6-benzhydryl-anilines were not accessible by conventional methods before. Our synthetic approach enables a high yield and a large-scale synthesis of this type of compound by a straightforward three-step alkylation/arylation procedure involving a non-catalysed substitution of the methoxy group using Grignard reagents. All anilines exhibit higher steric hindrance around the central nitrogen atom when compared to the published analogues as judged from the buried volumes. With this feature, they could be applied in many branches of synthetic chemistry.
We would like to acknowledge the financial support of the Czech Science Foundation (GA17-10377S) and European Social Fund (CZ.02.2.69/0.0/0.0/16_027/0008008).
Conflicts of interest
There are no conflicts to declare.Notes and references
- (a) P. P. Power, Nature, 2010, 463, 171–177 CrossRef CAS; (b) R. C. Fischer and P. P. Power, Chem. Rev., 2010, 110, 3877–3923 CrossRef CAS PubMed.
- For example see (a) L. Pu, B. T. Twamley and P. P. Power, J. Am. Chem. Soc., 2000, 122, 3524 CrossRef CAS; (b) T. J. Hadlington and C. Jones, Chem. Commun., 2014, 50, 2321–2323 RSC; (c) C. Jones, A. Sidiropoulos, N. Holzmann, G. Frenking and A. Stasch, Chem. Commun., 2012, 48, 9855–9857 RSC; (d) J. Li, C. Schenk, C. Goedecke, G. Frenking and C. Jones, J. Am. Chem. Soc., 2011, 133(46), 18622–18625 CrossRef CAS PubMed; (e) D. Gau, R. Rodriguez, T. Kato, N. Saffon-Merceron, A. de Cózar, F. P. Cossío and A. Baceiredo, Angew. Chem., Int. Ed., 2011, 50, 1092–1096 CrossRef CAS PubMed; (f) S. Khan, R. Michel, J. M. Dieterich, R. A. Mata, H. W. Roesky, J.-P. Demers, A. Lange and D. Stalke, J. Am. Chem. Soc., 2011, 133, 17889–17894 CrossRef CAS PubMed.
- (a) J. Huang, E. D. Stevens, S. P. Nolan and J. L. Petersen, J. Am. Chem. Soc., 1999, 121, 2674–2678 CrossRef CAS; (b) M. Scholl, T. M. Trnka, J. P. Morgan and R. H. Grubbs, Tetrahedron Lett., 1999, 40, 2247–2250 CrossRef CAS; (c) J. A. Love, J. P. Morgan, T. M. Trnka and R. H. Grubbs, Angew. Chem., Int. Ed., 2002, 41, 4035–4037 CrossRef CAS.
- For example see (a) M. N. Hopkinson, C. Richter, M. Schedler and F. Glorius, Nature, 2014, 510, 485–496 CrossRef CAS PubMed; (b) D. Janssen-Muller, C. Schlepphorst and F. Glorius, Chem. Soc. Rev., 2017, 46, 4845–4854 RSC; (c) M. Asay, C. Jones and M. Driess, Chem. Rev., 2011, 111, 354–396 CrossRef CAS PubMed; (d) L. Bourget-Merle, M. F. Lappert and J. R. Severn, Chem. Rev., 2002, 102, 3031–3065 CrossRef CAS PubMed; (e) C. Jones, Coord. Chem. Rev., 2010, 254, 1273–1289 CrossRef CAS.
- For example see (a) T. J. Hadlington, M. Hermann, G. Frenking and C. Jones, Chem. Sci., 2015, 6, 7249–7257 RSC; (b) T. J. Hadlington, M. Hermann, G. Frenking and C. Jones, J. Am. Chem. Soc., 2014, 136, 3028–3031 CrossRef CAS PubMed.
- (a) T. Beweries, R. Kuzora, U. Rozenthal, A. Schulz and A. Villinger, Angew. Chem., Int. Ed., 2011, 50, 8974–8978 CrossRef CAS PubMed; (b) A. Hinz, A. Schulz, A. Villinger and J.-M. Wolter, J. Am. Chem. Soc., 2015, 137, 3975–3980 CrossRef CAS PubMed.
- A. Hinz, A. Schulz and A. Villinger, J. Am. Chem. Soc., 2015, 137, 9953–9962 CrossRef CAS PubMed.
- (a) W.-J. Tao, R. Nakano, S. Ito and K. Nozaki, Angew. Chem., Int. Ed., 2016, 55, 2835–2839 CrossRef CAS PubMed; (b) A. Hinz and J. M. Goicoechea, Chem. – Eur. J., 2018, 24, 7358–7363 CrossRef CAS PubMed; (c) L. Liu, D. A. Ruiz, D. Munz and G. Bertrand, Chem, 2016, 1, 147–153 CrossRef CAS; (d) M. M. Hansmann, R. Jazzar and G. Bertrand, J. Am. Chem. Soc., 2016, 138, 8356–8359 CrossRef CAS PubMed.
- C. N. de Bruin-Dickason, A. J. Boutland, D. Dange, G. B. Deacon and C. Jones, Dalton Trans., 2018, 47, 9512–9520 RSC.
- J. Zhou, H.-F. Mao, L. Wang, J.-P. Zou and W. Zhang, Mol. Diversity, 2011, 15, 849–855 CrossRef CAS PubMed.
- E. Niecke, M. Nieger and F. Reichert, Angew. Chem., Int. Ed. Engl., 1988, 27, 1715–1716 CrossRef.
- (a) N. Burford, J. C. Landry, M. J. Ferguson and R. McDonald, Inorg. Chem., 2005, 44, 5897–5902 CrossRef CAS PubMed; (b) N. Burford, K. D. Conroy, J. C. Landry, P. J. Ragogna, M. J. Ferguson and R. McDonald, Inorg. Chem., 2004, 43, 8245–8251 CrossRef CAS PubMed; (c) N. Burford, T. S. Cameron, K. D. Conroy, B. Ellis, M. D. Lumsden, C. L. B. McDonald, R. McDonald, A. D. Phillips, P. J. Ragogna, R. W. Schurko, D. Walsh and R. E. Wasylishen, J. Am. Chem. Soc., 2002, 124, 14012–14013 CrossRef CAS PubMed; (d) N. Burford, J. A. C. Clyburne and M. S. W. Chan, Inorg. Chem., 1997, 36, 3204–3206 CrossRef CAS PubMed; (e) N. Burford, J. A. C. Clyburne, D. Silvert, S. Warner and W. A. Whitla, Inorg. Chem., 1997, 36, 482–484 CrossRef CAS.
- S. Meiries, G. L. Duc, A. Chartoire, A. Collado, K. Speck, K. S. A. Arachchige, A. M. Z. Slawin and S. P. Nolan, Chem. – Eur. J., 2013, 19, 17358–17368 CrossRef CAS PubMed.
- For example see (a) M. R. Harris, M. O. Konev and E. R. Jarvo, J. Am. Chem. Soc., 2014, 136, 7825–7828 CrossRef CAS PubMed; (b) P.-P. Chen, E. L. Lucas, M. A. Greene, S.-Q. Zhang, E. J. Tollefson, L. W. Erickson, B. L. H. Taylor, E. R. Jarvo and X. Hong, J. Am. Chem. Soc., 2019, 141, 5835–5855 CrossRef CAS PubMed; (c) B.-T. Guan, S.-K. Xiang, B.-Q. Wang, Z.-P. Sun, Y. Wang, K.-Q. Zhao and Z.-J. Shi, J. Am. Chem. Soc., 2008, 130, 3268–3269 CrossRef CAS PubMed.
- (a) E. Kumarasamy, R. Raghunathan, S. K. Kandappa, A. Sreenithya, S. Jockusch, R. B. Sunoj and J. Sivaguru, J. Am. Chem. Soc., 2017, 139, 655–662 CrossRef CAS PubMed; (b) E. Kumarasamy, R. Raghunathan, S. Jockusch, A. Ugrinov and J. Sivaguru, J. Am. Chem. Soc., 2014, 136, 8729–8737 CrossRef CAS PubMed; (c) E. Kumarasamy, R. Raghunathan, S. Jockusch, A. Ugrinov and J. Sivaguru, Chem. Commun., 2016, 52, 8305–8308 RSC; (d) L. Ye, K.-Y. Lo, Q. Gu and D. Yang, Org. Lett., 2017, 19, 308–311 CrossRef CAS PubMed.
- (a) F. G. Mann and F. H. C. Stewart, J. Am. Chem. Soc., 1954, 76, 2826–2832 RSC; (b) F. G. Mann and F. H. C. Stewart, Chem. Ind., 1954, 373 CAS.
- C. Görl and H. G. Alt, J. Mol. Catal. A: Chem., 2007, 273, 118–132 CrossRef.
- T. Maekawa, H. Sekizawa and K. Itami, Angew. Chem., Int. Ed., 2011, 50, 7022–7026 CrossRef CAS PubMed.
- (a) A. Poater, F. Ragone, S. Giudice, C. Costabile, R. Dorta, S. P. Nolan and L. Cavallo, Organometallics, 2008, 27, 2679–2681 CrossRef CAS; (b) A. Poater, B. Cosenza, A. Correa, S. Giudice, F. Ragone, V. Scarano and L. Cavallo, Eur. J. Inorg. Chem., 2009, 1759–1766 CrossRef CAS; (c) A. Poater, F. Ragone, R. Mariz, R. Dorta and L. Cavallo, Chem. – Eur. J., 2010, 16, 14348–14353 CrossRef CAS PubMed; (d) L. Falivene, R. Credendino, A. Poater, A. Petta, L. Serra, R. Oliva, V. Scarano and L. Cavallo, Organometallics, 2016, 35, 2286–2293 CrossRef CAS.
- Based on the search performed on November 17th 2019. CCDC - The Cambridge Structural Database. C. R. Groom, I. J. Bruno, M. P. Lightfoot and S. C. Ward, Acta Crystallogr., Sect. B: Struct. Sci., Cryst. Eng. Mater., 2016, 72, 171–179 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1956669–1956677. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c9cc09497k |
| This journal is © The Royal Society of Chemistry 2020 |