
One-pot aerobic oxidative sulfonamidation of aromatic thiols with ammonia by a dual-functional β-MnO2 nanocatalyst†
Keigo
Kamata  *a
and
*a
and
*a
and
Abstract
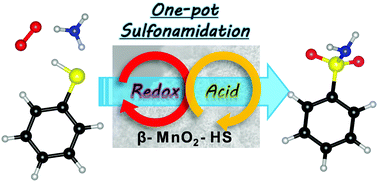
High-surface-area β-MnO2 (β-MnO2-HS) nanoparticles could act as effective heterogeneous catalysts for the one-pot oxidative sulfonamidation of various aromatic and heteroaromatic thiols to the corresponding sulfonamides using molecular oxygen (O2) and ammonia (NH3) as respective oxygen and nitrogen sources, without the need for any additives.
 Please wait while we load your content...
Please wait while we load your content...

