
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Acid promoted radical-chain difunctionalization of styrenes with stabilized radicals and (N,O)-nucleophiles†
Sensheng
Liu
and
Martin
Klussmann
 *
*
Max-Planck-Institut für Kohlenforschung, Kaiser-Wilhelm-Platz 1, 45470 Mülheim an der Ruhr, Germany. E-mail: klusi@mpi-muelheim.mpg.de
First published on 7th January 2020
Abstract
A difunctionalization of alkenes through sequential addition of a radical and a nucleophile has been developed, which is suggested to proceed by a radical chain mechanism not requiring a catalyst. An electron transfer step to the oxidant benzoyl peroxide is facilitated by protonation with a strong acid.
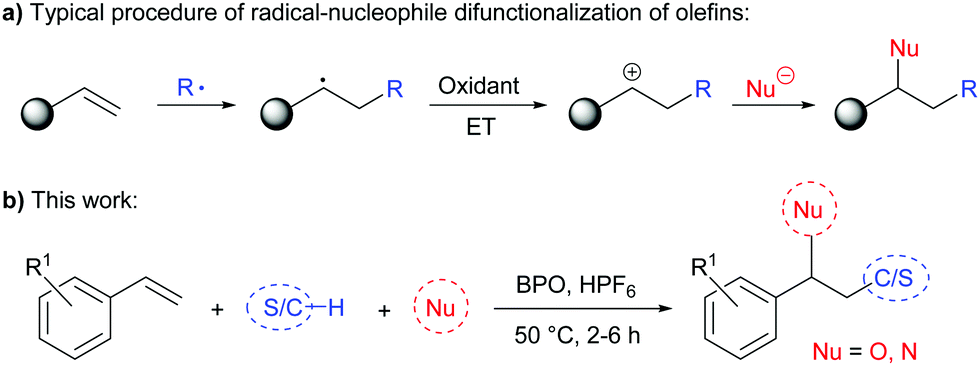
The difunctionalization of alkenes is a powerful transformation in synthetic organic chemistry. Besides transition-metal catalysed methods that proceed via organometallic intermediates,1 such reactions can be efficiently conducted by addition of free radicals.2 An interesting strategy amongst these is the consecutive addition of a radical and a nucleophile, which requires an electron transfer (ET) step after the radical addition, in order to generate a carbocation that could be trapped by a nucleophile (Scheme 1a).2a,3 Such reactions would enable functionalizing olefins with a wide variety of reagents in a regioselective manner, given that radical precursors and nucleophiles mostly react complementarily. Although many synthetically interesting methods have been developed towards this goal, there is as of yet no method with a truly broad substrate scope of both radicals and nucleophiles.4–6 Most of these methods require the presence of a transition metal catalyst or reagent to achieve the desired ET forming the carbocation intermediate, notable exceptions utilize an organic photocatalyst,7 iodide as catalyst8 or electrochemistry.9
 | ||
| Scheme 1 Radical-nucleophile addition of olefins. BPO = Benzoyl peroxide. | ||
We had previously worked on the activation of tert-butyl hydroperoxide by Brønsted acids, most notably in the presence of ketones.10 We noticed the work by Zhang, Bao and co-workers, who reported a copper-catalysed difunctionalization of alkenes using benzoyl peroxide (BPO) in the presence of HPF6.11 Acetonitrile was both radical precursor and nucleophile and the role of the acid was not clear, thus it raised our interest for its combination of a peroxide and acid and its potential to add radicals and nucleophiles to olefins.
Here, we report mechanistic details of the effect of acid on benzoyl peroxide and a method for difunctionalization of styrene derivatives with stabilized C- and S-radicals and N- and O-nucleophiles. The reactions do not require a catalyst but the presence of a strong Brønsted acid, and they operate at only slightly elevated temperature (Scheme 1b).
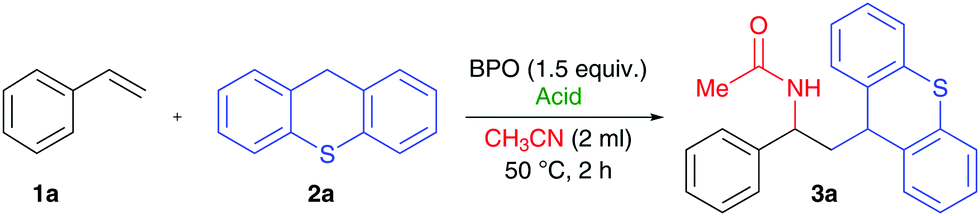
We found that the combination of BPO with HPF6 allowed for the addition of thioxanthene (2a) and acetonitrile to styrene (1a) without any additional catalyst within two hours at 50 °C (Table 1, entry 1). The product's structure (3a) suggested that a thioxanthenyl radical was added to styrene and subsequently acetonitrile attacked as a nucleophile in a Ritter reaction.12 The C-radical of thioxanthene had apparently formed by H-atom transfer (HAT),13 presumably to a benzoyloxyl radical generated from BPO.
|
|
|||
|---|---|---|---|
| Entry | Acid | Acid equiv. | Yieldb (%) |
| a 1a (0.2 mmol), 2a (0.4 mmol, 2.0 equiv.), BPO (0.3 mmol, 1.5 equiv.), Acid (0.2 mmol, 1.0 equiv.) in CH3CN (2 ml). b Yields determined by 1H NMR spectroscopic analysis of the crude reaction mixture relative to internal standard CH3NO2, yield of isolated product in parentheses. c With addition of 1.0 equiv. of water. d Degassed, under argon. e Performed on a larger scale, isolating 1.5 g of 3a. | |||
| 1 | HPF6 (aq., 55%) | 1.0 | 88 |
| 2 | HPF6 (aq., 55%)c | 0.1 | 20 |
| 3 | — | — | 0 |
| 4 | CF3CO2Hc | 1.0 | 11 |
| 5 | HBF4 (aq., 48%) | 1.0 | 39 |
| 6 | HClO4 (aq., 70%) | 1.0 | 51 |
| 7d | HPF6 (aq., 55%) | 1.0 | 91 (88, 83e) |
The acid plays a crucial role for the reaction: with lower amounts, the yield drops significantly (entry 2) and without acid, no reaction occurs (entry 3; for further results under changed reaction conditions, see the ESI†). In the presence of other acids, the product was also formed, but apparently the yield is correlated with the acid strength. For example, trifluoroacetic acid gave only 11% of 3a, while the stronger acids HBF4 and HClO4 gave 39% and 51% (entries 4–6). In the absence of BPO and with other peroxide oxidants, the product was not formed. Ambient temperature is sufficient for the reaction, but the rate is significantly reduced. Performing the reaction under strict exclusion of oxygen increased the yield, and the reaction could also be performed on a larger scale, giving 1.5 g of 3a with an isolated yield of 83% (entry 7).
The reaction is very likely proceeding via a radical mechanism, as the addition of radical inhibitors reduced the yield significantly (see the ESI† for details). The acid apparently does not affect the decomposition of BPO, which has a reported 10 hour half-life temperature of 73 °C.14 As an NMR experiment revealed, BPO with or without acid did not change when heated in acetonitrile at 50 °C for two hours (Scheme 2a). However, in the presence of thioxanthene, benzoic acid was formed in significant amounts under these conditions, indicating that it accelerates the peroxide decomposition (Scheme 2b).
 | ||
| Scheme 2 Experiments pointing to the reaction mechanism. | ||
While the acid does not accelerate the homolytic cleavage of BPO, it does change its redox potential. We studied this effect by cyclic voltammetry (Fig. 1). The reduction of BPO alone was found to occur at −345 mV, which underwent a shift by +470 mV in the presence of 0.66 equiv. of HPF6, the relative amount used under reaction conditions. Other acids also induced such a shift, but less strong, as is shown here for trifluoroacetic acid (for other acids, see the ESI†). The reduction was thus significantly eased by the strong acid HPF6, possibly by protonation that turns the now cationic peroxide into a better electron acceptor.
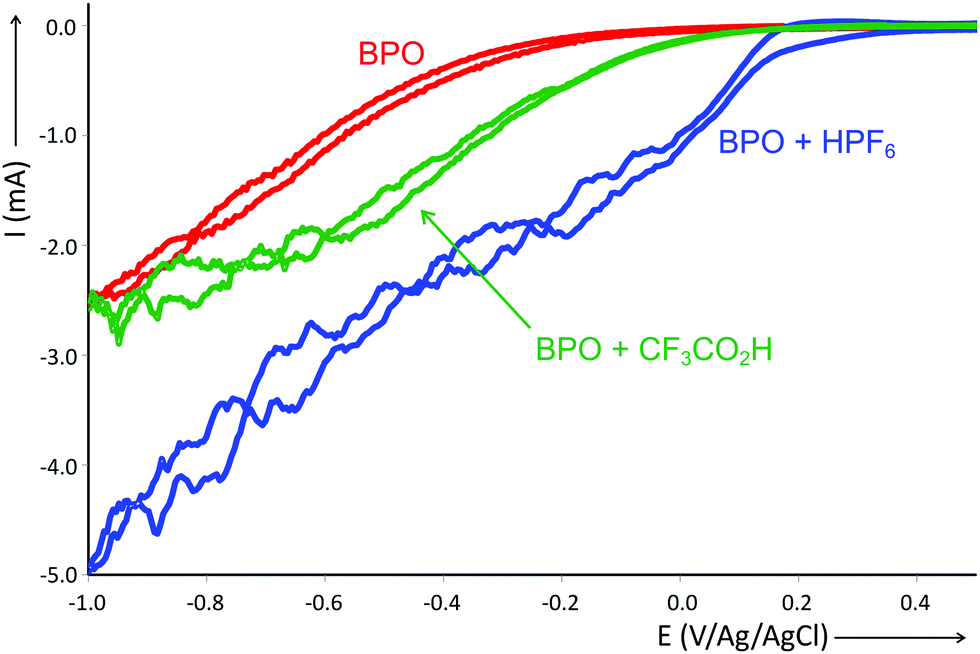 | ||
| Fig. 1 Cyclic voltammograms showing the effect of acid addition on the reduction potential of BPO. Two platinized Pt wires as a counter and working electrode with a Ag/AgCl electrode as a reference were used. The cyclic voltammetry (CV) was conducted from −1.0 V to 2.0 V with a scan rate of 100 mV s−1. BPO (0.3 mmol), acid (0.2 mmol), tetrabutylammonium hexafluorophosphate (0.1 M) in CH3CN under Ar. | ||
These results indicate a reaction mechanism that relies on electron transfer (ET) steps (Scheme 3). Initiating benzoyloxyl radicals (4) are formed from BPO in the presence of thioxanthene, possibly by ET to BPO that is facilitated by protonation. These induce HAT from thioxanthene, generating a new radical (5), which then adds to styrene, forming the benzylic radical 6. This is oxidized by BPO in the presence of HPF6, most likely by ET to the protonated peroxide (7), giving the intermediate carbocation 8, benzoate and a new benzoyloxyl radical. The cation 8 can react as an electrophile with acetonitrile, generating the product 3a in the fashion of a Ritter reaction. Thus, the reaction appears to run by a radical chain mechanism and can be seen as a case of “electron-catalysis”.15
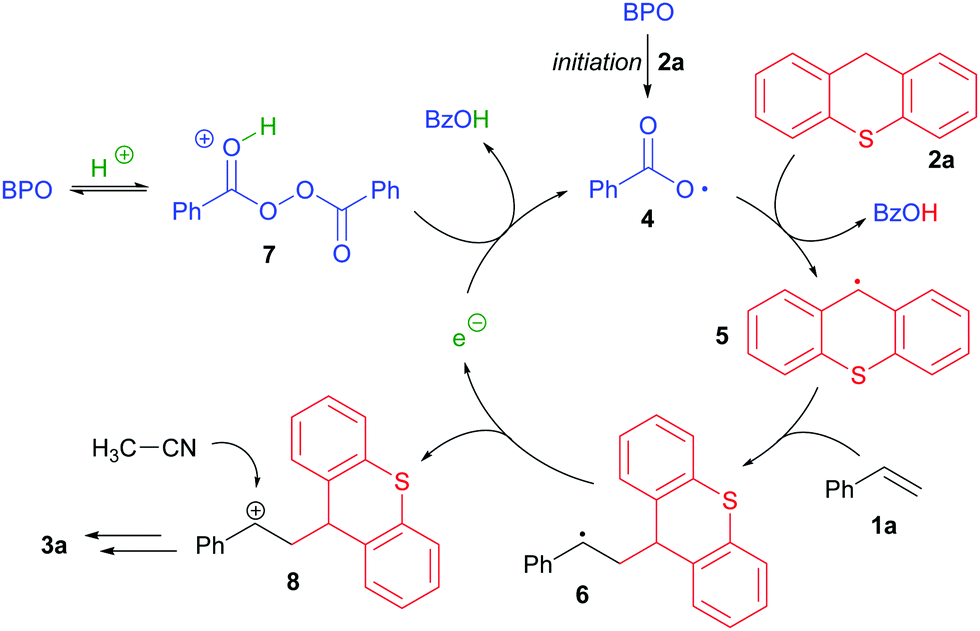 | ||
| Scheme 3 Potential reaction mechanism, shown exemplary for 1a and 2a. | ||
Based on this working model of the reaction's mechanism, other substrates that can initiate such a radical chain by interaction with BPO16 and that easily form radicals by HAT to a benzoyloxyl radical should also be employable, as well as other olefins and alternative nucleophiles.
As shown in Scheme 4, styrenes with both weakly electron-donating (Me, tBu) and withdrawing (F, Cl, Br) substituents on the aromatic ring, regardless of their positions, afforded the desired products in good yields 62–96% (3b–3f, 3j–3k and 3m–3o), as did 4-vinylbiphenyl and vinylnaphthalene (3i, 3p). However, styrene bearing the strongly electron-donating methoxy substituent did not give the desired product, and the strongly electron-withdrawing NO2 and CF3 substituents led to low yields of 3g, 3h and 3l in 10%, 30% and 33%. Using indene as olefin gave the product 3q in 30% yield, but it is remarkable for its high trans-selectivity. A diastereomeric ratio of >21![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 was determined in the crude reaction mixture, but after purification, we received the pure trans-product 3q. Similarly, only the trans-product 3r was isolated from the reaction with E-β-methylstyrene. Strangely, other nitriles besides acetonitrile did not lead to the expected products with thioxanthene. However, when we used xanthene as HAT-donor, we could isolate different amide products by performing the reaction in different nitriles as solvent. Aliphatic and aromatic nitriles as well gave the products 3s–3x with good yields after an extended reaction time of 6 hours. The general structure of these products was confirmed by X-ray crystallography of product 3e.
:1 was determined in the crude reaction mixture, but after purification, we received the pure trans-product 3q. Similarly, only the trans-product 3r was isolated from the reaction with E-β-methylstyrene. Strangely, other nitriles besides acetonitrile did not lead to the expected products with thioxanthene. However, when we used xanthene as HAT-donor, we could isolate different amide products by performing the reaction in different nitriles as solvent. Aliphatic and aromatic nitriles as well gave the products 3s–3x with good yields after an extended reaction time of 6 hours. The general structure of these products was confirmed by X-ray crystallography of product 3e.
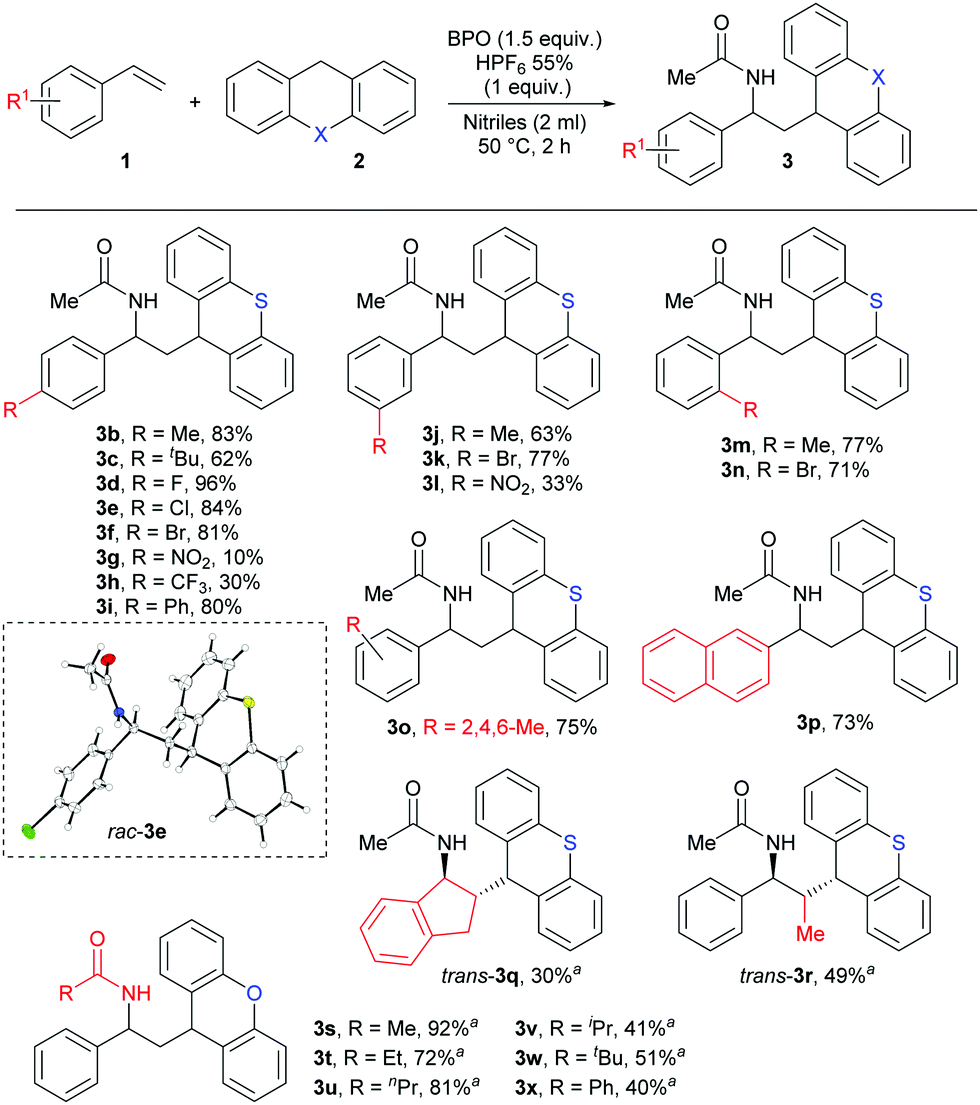 | ||
| Scheme 4 Substrate scope for the reaction of styrenes and nitriles: 1 (0.2 mmol), 2 (0.4 mmol, 2.0 equiv.), BPO (0.3 mmol, 1.5 equiv.), HPF6 (0.2 mmol, 1.0 equiv.) and nitriles (2 ml), Ar, isolated yield. aReaction time: 6 hours. ORTEP diagram is drawn with displacement ellipsoids at the 50% probability level. | ||
Next, the scope with respect to nucleophiles was explored. Although we tried many substrates (see the ESI† for further details), only alcohols were successful, and only with thioxanthene but not with xanthene (Scheme 5). Reactions of styrene with various alcohols produced the expected products in good yields, with primary alcohols in generally higher yields (9a–9e, 73–93%) than secondary (9f–9g) and tertiary alcohols (9h).
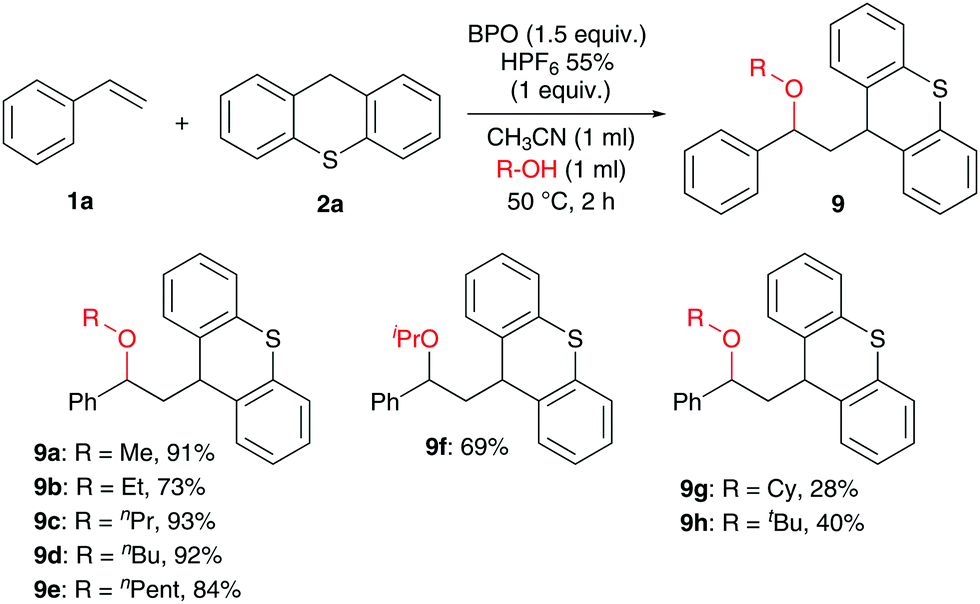 | ||
| Scheme 5 Substrate scope for the reaction of alcohol nucleophiles: 1a (0.2 mmol), 2a (0.4 mmol, 2.0 equiv.), BPO (0.3 mmol, 1.5 equiv.), HPF6 (0.2 mmol, 1.0 equiv.), alcohols (1 ml) and CH3CN (1 ml), Ar, isolated yield. | ||
Thiophenols (10) as HAT-donors with acetonitrile as nucleophile could also be employed successfully in this reaction with styrene (Scheme 6). While alkyl thiols did not react under those conditions, products 11 with various differently substituted thiophenols could be employed. Products of a thiol–ene reaction were not observed. Similar products like 11 had recently been reported, being synthesized by an iodide-catalysed radical reaction8 or by ionic reactions also utilizing stoichiometric amounts of oxidants.17
 | ||
| Scheme 6 Substrate scope for thiylation: 1a (0.2 mmol), 10 (0.4 mmol, 2.0 equiv.), BPO (0.3 mmol, 1.5 equiv.), HPF6 (0.2 mmol, 1.0 equiv.) and CH3CN (2 ml), Ar, isolated yield. | ||
Substrates not capable of initiating BPO decomposition obviously fail in this reaction. However, addition of extra initiators may overcome this limitation. We found that addition of N,N-dimethylanilines, well-known initiators for BPO,18 enable the addition of two molecules of acetonitrile to styrene, furnishing 13 (Scheme 7). Although the yields are not as high as with Cu-catalysts,11 48% is reached with the use of 10 mol% of the p-bromo aniline. The product yield is obviously linked to the initiation rate and electronic properties of the anilines, as the comparison with more and less electron rich derivatives shows.
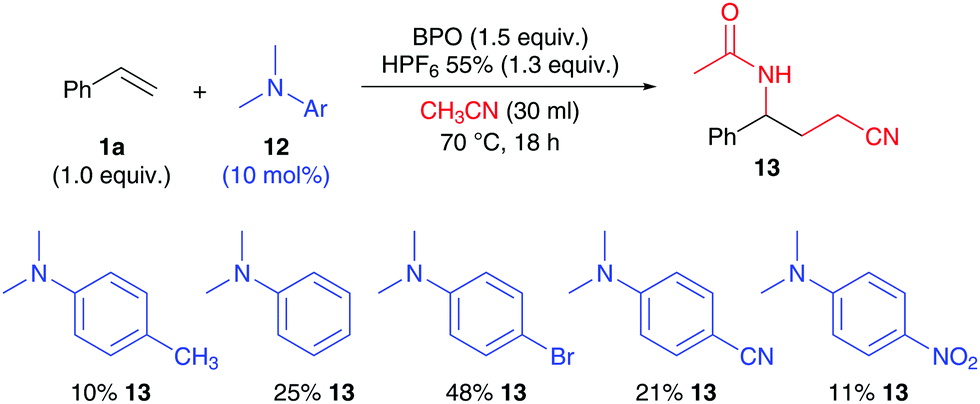 | ||
| Scheme 7 Investigating dimethylaniline initiators: 1a (0.5 mmol), BPO (0.75 mmol, 1.5 equiv.), HPF6 (0.66 mmol, 1.3 equiv.), 12 (0.05 mmol) and CH3CN (30 ml), isolated yield. | ||
In conclusion, a method for the difunctionalization of styrenes with radicals derived from thioxanthene, xanthene and thiophenols together with nitrile and alcohol nucleophiles was developed. The combination of benzoyl peroxide with HPF6, a strong Brønsted acid, is a key element of the reaction that does not require transition-metal catalysts, high temperatures or prolonged reaction times. Mechanistic studies suggest that the acid can promote the electron transfer to the peroxide, and that the reaction proceeds by a radical chain that is initiated by interaction of the radical precursor with the peroxide. Addition of an extra radical initiator can overcome this limitation, which suggests a way to extend this synthetic strategy.
M. K. thanks the DFG (KL 2221/4-2, Heisenberg scholarship) and S. L. thanks the China Scholarship Council (CSC, doctoral scholarship No. 201808420290). We are grateful for the help of the analytical departments of the MPI für Kohlenforschung and for help in cyclic voltammetry measurements by Dr Yuxiao Ding (MPI for Chemical Energy Conversion). Open Access funding provided by the Max Planck Society.
Conflicts of interest
There are no conflicts to declare.Notes and references
- For a review, see: (a) J.-S. Zhang, L. Liu, T. Chen and L.-B. Han, Chem. – Asian J., 2018, 13, 2277 CrossRef CAS PubMed; (b) F. Wang, X. Qi, Z. Liang, P. Chen and G. Liu, Angew. Chem., Int. Ed., 2014, 53, 1881 CrossRef CAS PubMed; (c) A. Bunescu, Q. Wang and J. Zhu, Angew. Chem., Int. Ed., 2015, 54, 3132 CrossRef CAS PubMed.
- (a) X.-W. Lan, N.-X. Wang and Y. Xing, Eur. J. Org. Chem., 2017, 5821 CrossRef CAS; (b) H. Yi, G. Zhang, H. Wang, Z. Huang, J. Wang, A. K. Singh and A. Lei, Chem. Rev., 2017, 117, 9016 CrossRef CAS PubMed; (c) H. Egami and M. Sodeoka, Angew. Chem., Int. Ed., 2014, 53, 8294 CrossRef CAS PubMed; (d) H. Fischer and L. Radom, Angew. Chem., Int. Ed., 2001, 40, 1340 CrossRef CAS.
- F. Minisci, Acc. Chem. Res., 1975, 8, 165 CrossRef CAS.
- For an overview of early methods using stoichiometric Mn(OAc)3, see: G. G. Melikyan, Synthesis, 1993, 833 CrossRef CAS.
- For C-radicals, see: (a) P. G. Janson, I. Ghoneim, N. O. Ilchenko and K. J. Szabó, Org. Lett., 2012, 14, 2882 CrossRef CAS PubMed; (b) H. Egami, R. Shimizu and M. Sodeoka, Tetrahedron Lett., 2012, 53, 5503 CrossRef CAS; (c) Y. Yasu, T. Koike and M. Akita, Org. Lett., 2013, 15, 2136 CrossRef CAS PubMed; (d) A. Carboni, G. Dagousset, E. Magnier and G. Masson, Org. Lett., 2014, 16, 1240 CrossRef CAS PubMed; (e) H. Yi, X. Zhang, C. Qin, Z. Liao, J. Liu and A. Lei, Adv. Synth. Catal., 2014, 356, 2873 CrossRef CAS; (f) C. Chatalova-Sazepin, Q. Wang, G. M. Sammis and J. Zhu, Angew. Chem., Int. Ed., 2015, 54, 5443 CrossRef CAS PubMed; (g) Y.-Y. Liu, X.-H. Yang, R.-J. Song, S. Luo and J.-H. Li, Nat. Commun., 2017, 8, 14720 CrossRef PubMed; (h) B. Qian, S. Chen, T. Wang, X. Zhang and H. Bao, J. Am. Chem. Soc., 2017, 139, 13076 CrossRef CAS PubMed; (i) S. N. Gockel, T. L. Buchanan and K. L. Hull, J. Am. Chem. Soc., 2018, 140, 58 CrossRef CAS PubMed; (j) W. Deng, W. Feng, Y. Li and H. Bao, Org. Lett., 2018, 20, 4245 CrossRef CAS PubMed; (k) R. Su, Y. Li, M.-Y. Min, X.-H. Ouyang, R.-J. Song and J.-H. Li, Chem. Commun., 2018, 54, 13511 RSC; (l) Y.-X. Dong, Y. Li, C.-C. Gu, S.-S. Jiang, R.-J. Song and J.-H. Li, Org. Lett., 2018, 20, 7594 CrossRef CAS PubMed; (m) X.-H. Ouyang, Y. Li, R.-J. Song, M. Hu, S. Luo and J.-H. Li, Sci. Adv., 2019, 5, eaav9839 CrossRef CAS PubMed.
- For heteroatom-radicals, see: (a) Y. Gao, X. Li, W. Chen, G. Tang and Y. Zhao, J. Org. Chem., 2015, 80, 11398 CrossRef CAS PubMed; (b) Y. Yang, R.-J. Song, X.-H. Ouyang, C.-Y. Wang, J.-H. Li and S. Luo, Angew. Chem., Int. Ed., 2017, 56, 7916 CrossRef CAS PubMed; (c) B. Du, Y. Wang, H. Mei, J. Han and Y. Pan, Adv. Synth. Catal., 2017, 359, 1684 CrossRef CAS; (d) Y. Wang, W. Wang, R. Tang, Z. Liu, W. Tao and Z. Fang, Org. Biomol. Chem., 2018, 16, 7782 RSC.
- N. Noto, T. Koike and M. Akita, Chem. Sci., 2017, 8, 6375 RSC.
- Y. Zheng, Y. He, G. Rong, X. Zhang, Y. Weng, K. Dong, X. Xu and J. Mao, Org. Lett., 2015, 17, 5444 CrossRef CAS PubMed.
- Y. Yuan, Y. Cao, Y. Lin, Y. Li, Z. Huang and A. Lei, ACS Catal., 2018, 8, 10871 CrossRef CAS.
- (a) B. Schweitzer-Chaput, A. Sud, Á. Pinter, S. Dehn, P. Schulze and M. Klussmann, Angew. Chem., Int. Ed., 2013, 52, 13228 CrossRef CAS PubMed; (b) B. Schweitzer-Chaput, J. Demaerel, H. Engler and M. Klussmann, Angew. Chem., Int. Ed., 2014, 53, 8737 CrossRef CAS PubMed; (c) B. Schweitzer-Chaput, T. Kurtén and M. Klussmann, Angew. Chem., Int. Ed., 2015, 54, 11848 CrossRef CAS PubMed; (d) H.-L. Yue and M. Klussmann, Synlett, 2016, 2505 CAS; (e) B. Schweitzer-Chaput, E. Boess and M. Klussmann, Org. Lett., 2016, 18, 4944 CrossRef CAS PubMed; (f) W. Shao, M. Lux, M. Breugst and M. Klussmann, Org. Chem. Front., 2019, 6, 1796 RSC.
- N. Zhu, T. Wang, L. Ge, Y. Li, X. Zhang and H. Bao, Org. Lett., 2017, 19, 4718 CrossRef CAS PubMed.
- (a) B. H. Hoff, Synthesis, 2018, 2824 CrossRef CAS; (b) D. Jiang, T. He, L. Ma and Z. Wang, RSC Adv., 2014, 4, 64936 RSC.
- (a) M. Milan, M. Salamone, M. Costas and M. Bietti, Acc. Chem. Res., 2018, 51, 1984 CrossRef CAS PubMed; (b) T. V. RajanBabu and F. Gagosz, in Encyclopedia of Reagents for Organic Synthesis, 2005, DOI:10.1002/047084289X.rd022.pub2.
- C. S. Sheppard, Encyclopedia of polymer science and engineering, John Wiley & Sons, Inc., 1985, vol. 11, p. 1 Search PubMed.
- A. Studer and D. P. Curran, Nat. Chem., 2014, 6, 765 CrossRef CAS PubMed.
- The induced decomposition of BPO could also be shown for the other radical precursors used in this study, see the ESI† for details.
- (a) A. Bewick, J. M. Mellor and W. M. Owton, J. Chem. Soc., Perkin Trans. 1, 1985, 1039 RSC; (b) L. Benati, P. C. Montevecchi and P. Spagnolo, J. Chem. Soc., Perkin Trans. 1, 1987, 2815 RSC; (c) H. Cui, X. Liu, W. Wei, D. Yang, C. He, T. Zhang and H. Wang, J. Org. Chem., 2016, 81, 2252 CrossRef CAS PubMed; (d) D. Wang, Z. Yan, Q. Xie, R. Zhang, S. Lin and Y. Wang, Org. Biomol. Chem., 2017, 15, 1998 RSC.
- (a) L. Horner and E. Schwenk, Liebigs Ann. Chem., 1950, 566, 69 CrossRef CAS; (b) A. Székely and M. Klussmann, Chem. – Asian J., 2019, 14, 105 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Including experimental details, NMR spectra, X-ray crystallographic data and a CIF file. CCDC 1957001. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c9cc09369a |
| This journal is © The Royal Society of Chemistry 2020 |